ZEPOSIA (ozanimod hydrochloride 0.92 mg) Dailymed
Generic: ozanimod hydrochloride is used for the treatment of Angina, Unstable Ischemic Attack, Transient Heart Failure Myocardial Infarction Sick Sinus Syndrome Sinoatrial Block Sleep Apnea Syndromes Stroke Multiple Sclerosis, Relapsing-Remitting Atrioventricular Block

Go PRO for all pill images
Recent Major Changes Section
Dosage and Administration (2.2 ,2.3 )Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â 8/2023
Warnings and Precautions (5.2 ,5.4 ,5.11 )Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â 8/2023
1 Indications And Usage
ZEPOSIA is indicated for the treatment of:
• relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.• moderately to severely active ulcerative colitis (UC) in adults.
ZEPOSIA is a sphingosine 1-phosphate receptor modulator indicated for the treatment of:
• Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. (1 )• Moderately to severely active ulcerative colitis (UC) in adults. (1 )
2 Dosage And Administration
• Assessments are required prior to initiating ZEPOSIA. (2.1 )• Titration is required for treatment initiation. (2.2 )• The recommended maintenance dosage is 0.92 mg orally once daily. (2.2 )• The recommended maintenance dosage in patients with mild or moderate chronic hepatic impairment (Child-Pugh class A or B) is 0.92 mg once every other day. (2.3 )• If a dose is missed within the first 2 weeks of treatment, reinitiate with the titration regimen. If a dose is missed after the first 2 weeks of treatment, continue treatment as planned. (2.4 )2.1 Assessments Prior to First Dose of ZEPOSIA
Before initiation of treatment with ZEPOSIA, assess the following:
Complete Blood Count
Obtain a recent (i.e., within the last 6 months or after discontinuation of prior MS or UC therapy) complete blood count (CBC), including lymphocyte count [see Warnings and Precautions (5.1)].
Cardiac Evaluation
Obtain an electrocardiogram (ECG) to determine whether preexisting conduction abnormalities are present. In patients with certain preexisting conditions, advice from a cardiologist should be sought [see Warnings and Precautions (5.3) ].
Liver Function Tests
Obtain recent (i.e., within the last 6 months) transaminase and bilirubin levels [see Warnings and Precautions (5.4) ].
Ophthalmic Assessment
In patients with a history of uveitis or macular edema, obtain an evaluation of the fundus, including the macula [see Warnings and Precautions (5.8) ].
Current or Prior Medications
• If patients are taking anti-neoplastic, non-corticosteroid immunosuppressive, or immune-modulating therapies, or if there is a history of prior use of these drugs, consider possible unintended additive immunosuppressive effects before initiating treatment with ZEPOSIA [see Warnings and Precautions (5.1) and Drug Interactions (7)].• Determine if patients are taking drugs that could slow heart rate or atrioventricular conduction [see Warnings and Precautions (5.3) and Drug Interactions (7)].
Vaccinations
Patients without a healthcare professional-confirmed history of chickenpox or without documentation of a full course of vaccination against varicella zoster virus (VZV) should be tested for antibodies to VZV before initiating ZEPOSIA; VZV vaccination of antibody-negative patients is recommended prior to commencing treatment with ZEPOSIA [see Warnings and Precautions (5.1) Â and Drug Interactions (7)].
If live attenuated vaccine immunizations are required, administer at least 1 month prior to initiation of ZEPOSIA.
2.2 Recommended Dosage for Multiple Sclerosis and Ulcerative Colitis
Initiate ZEPOSIA with a 7-day titration, as shown in Table 1 [see Warnings and Precautions (5.3) ]. After initial titration, the recommended dosage of ZEPOSIA is 0.92 mg taken orally once daily starting on Day 8.
Swallow ZEPOSIA capsules whole, with or without food [see Clinical Pharmacology (12.3)].
Table 1: Dose Titration Regimen
Days 1-4
0.23 mg once daily
Days 5-7
0.46 mg once daily
Day 8 and thereafter
0.92 mg once daily*
*Patients with mild to moderate hepatic impairment (Child-Pugh class A or B) should take 0.92 mg once every other day [see Recommended Dosage in Patients with Hepatic Impairment (2.3).]
2.3 Recommended Dosage in Patients with Hepatic Impairment
In patients with mild or moderate hepatic impairment (Child-Pugh class A or B), initiate ZEPOSIA with a 7-day titration, as shown in Table 1. After initial titration, the recommended dosage of ZEPOSIA in these patients is 0.92 mg taken orally once every other day, starting on Day 8 [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Reinitiation of ZEPOSIA after Treatment Interruption
If a dose of ZEPOSIA is missed during the first 2 weeks of treatment, reinitiate treatment using the titration regimen [see Dosage and Administration (2.2)].
If a dose of ZEPOSIA is missed after the first 2 weeks of treatment, continue with the treatment as planned.
3 Dosage Forms And Strengths
Capsules:
• 0.23 mg ozanimod: light grey opaque body/light grey opaque cap imprinted with black ink "OZA" on the cap and "0.23 mg" on the body• 0.46 mg ozanimod: light grey opaque body/orange opaque cap imprinted with black ink "OZA" on the cap and "0.46 mg" on the body• 0.92 mg ozanimod: orange opaque body/orange opaque cap imprinted with black ink "OZA" on the cap and "0.92 mg" on the body
Capsules: 0.23 mg, 0.46 mg, 0.92 mg ozanimod (3 )
4 Contraindications
ZEPOSIA is contraindicated in patients who:
• In the last 6 months, have experienced a myocardial infarction, unstable angina, stroke, transient ischemic attack (TIA), decompensated heart failure requiring hospitalization, or Class III or IV heart failure [see Warnings and Precautions (5.3) ]• Have the presence of Mobitz type II second-degree or third degree atrioventricular (AV) block, sick sinus syndrome, or sino-atrial block, unless the patient has a functioning pacemaker [see Warnings and Precautions (5.3)]• Have severe untreated sleep apnea [see Warnings and Precautions (5.3)]• Are taking a monoamine oxidase (MAO) inhibitor [see Drug Interactions (7)]
• In the last 6 months, experienced myocardial infarction, unstable angina, stroke, transient ischemic attack, decompensated heart failure requiring hospitalization, or Class III or IV heart failure. (4 )• Presence of Mobitz type II second-degree or third degree atrioventricular (AV) block, sick sinus syndrome, or sino-atrial block, unless the patient has a functioning pacemaker. (4 )• Severe untreated sleep apnea. (4 )• Concomitant use of a monoamine oxidase inhibitor. (4 ,7 )
5 Warnings And Precautions
• Infections: ZEPOSIA may increase the risk of infections. Obtain a complete blood count (CBC) before initiation of treatment. Monitor for infection during treatment and for 3 months after discontinuation. Do not start ZEPOSIA in patients with active infections. (5.1 )• Progressive Multifocal Leukoencephalopathy (PML): Withhold ZEPOSIA at the first sign or symptom suggestive of PML. (5.2 )• Bradyarrhythmia and Atrioventricular Conduction Delays: ZEPOSIA may result in transient decrease in heart rate; titration is required for treatment initiation. Check an electrocardiogram (ECG) to assess for preexisting cardiac conduction abnormalities before starting ZEPOSIA. Consider cardiology consultation for conduction abnormalities or concomitant use with other drugs that decrease heart rate. (2.1 ,2.2 ,5.3 ,7 )• Liver Injury: Discontinue if significant liver injury is confirmed. Obtain liver function tests before initiating ZEPOSIA. (5.4 )• Fetal Risk: Women of childbearing potential should use effective contraception during treatment and for 3 months after stopping ZEPOSIA. (5.5 ,8.3 )• Increased Blood Pressure (BP): Monitor BP during treatment. (5.6 )• Respiratory Effects: May cause a decline in pulmonary function. Assess pulmonary function (e.g., spirometry) if clinically indicated. (5.7 )• Macular Edema: A prompt ophthalmic evaluation is recommended if there is any change in vision while taking ZEPOSIA. Diabetes mellitus and uveitis increase the risk of macular edema; patients with a history of these conditions should have an ophthalmic evaluation of the fundus, including the macula, prior to treatment initiation. (5.8 )5.1 Infections
Risk of Infections
ZEPOSIA causes a mean reduction in peripheral blood lymphocyte count to approximately 45% of baseline values because of reversible sequestration of lymphocytes in lymphoid tissues [see Clinical Pharmacology (12.2)]. ZEPOSIA may therefore increase the susceptibility to infections, some serious in nature. Life-threatening and rare fatal infections have occurred in patients receiving ZEPOSIA.
Obtain a recent (i.e., within 6 months or after discontinuation of prior MS or UC therapy) complete blood count (CBC) including lymphocyte count before initiation of ZEPOSIA.
Delay initiation of ZEPOSIA in patients with an active infection until the infection is resolved.
In MS Study 1 and Study 2, the overall rate of infections and rate of serious infections in patients treated with ZEPOSIA were similar to that in patients who received interferon (IFN) beta-1a (35% vs. 34% and 1% vs. 0.8%, respectively). In UC Study 1 and Study 3, the overall rate of infections and rate of serious infections in patients treated with ZEPOSIA were similar to that in patients who received placebo (9.9% vs. 10.7% and 0.8% vs. 0.4%, respectively). In UC Study 2, the overall rate of infections in patients treated with ZEPOSIA was higher than in patients treated with placebo (23% vs. 12%) and the rate of serious infections was similar (0.9% vs. 1.8%).
ZEPOSIA increased the risk of viral upper respiratory tract infections, urinary tract infections, and herpes infections [see Adverse Reactions (6.1)].
The proportion of patients treated with ZEPOSIA who experienced lymphocyte counts less than 0.2 Ă— 109/L was 3.3% in MS Study 1 and Study 2. The proportion of patients treated with ZEPOSIA with lymphocyte counts less than 0.2 Ă— 109/L was 2% in UC Study 1 and Study 3 and 2.3% in UC Study 2. These values generally returned to greater than 0.2 Ă— 109/L while patients remained on treatment with ZEPOSIA. After discontinuing ZEPOSIA 0.92 mg, the median time for peripheral blood lymphocytes to return to the normal range was approximately 30 days, with approximately 80% to 90% of patients in the normal range within 3 months [see Clinical Pharmacology (12.2)].
Consider interruption of treatment with ZEPOSIA if a patient develops a serious infection.
Because the elimination of ZEPOSIA after discontinuation may take up to 3 months, continue monitoring for infections throughout this period.
Herpes Viral Infection
Cases of localized herpes virus infection (e.g., herpes zoster and herpes simplex) were seen in clinical trials of ZEPOSIA.
In MS Study 1 and Study 2, herpes zoster was reported as an adverse reaction in 0.6% of patients treated with ZEPOSIA 0.92 mg and in 0.2% of patients who received IFN beta-1a.
In UC Study 1 and Study 3, herpes zoster was reported in 0.4% of patients who received ZEPOSIA and none in patients who received placebo. In UC Study 2, herpes zoster was reported in 2.2% of patients who received ZEPOSIA and 0.4% of patients who received placebo. None were serious or disseminated.
Herpes simplex encephalitis and varicella zoster meningitis have been reported with sphingosine 1-phosphate (S1P) receptor modulators. Patients without a healthcare professional-confirmed history of varicella (chickenpox), or without documentation of a full course of vaccination against varicella zoster virus (VZV), should be tested for antibodies to VZV before initiating ZEPOSIA (see Vaccinations below).
Cryptococcal Infection
Cases of fatal cryptococcal meningitis (CM) and disseminated cryptococcal infections have been reported with S1P receptor modulators. Physicians should be vigilant for clinical symptoms or signs of CM. Patients with symptoms or signs consistent with a cryptococcal infection should undergo prompt diagnostic evaluation and treatment. ZEPOSIA treatment should be suspended until a cryptococcal infection has been excluded. If CM is diagnosed, appropriate treatment should be initiated.
Prior and Concomitant Treatment with Anti-Neoplastic, Non-Corticosteroid Immunosuppressive, or Immune-modulating Therapies
In the MS and UC clinical studies, patients who received ZEPOSIA were not to receive concomitant treatment with anti-neoplastic, non-corticosteroid immunosuppressive, or immune-modulating therapies used for the treatment of MS and UC. Concomitant use of ZEPOSIA with any of these therapies would be expected to increase the risk of immunosuppression. In UC studies, concomitant use of corticosteroids was allowed and did not appear to influence the safety or efficacy of ZEPOSIA [see Clinical Studies (14.2)].
Anti-neoplastic, immune-modulating, or immunosuppressive therapies (including corticosteroids) should be co-administered with caution because of the risk of additive immune system effects during such therapy. When switching to ZEPOSIA from immunosuppressive medications, consider the duration of their effects and their mode of action to avoid unintended additive immunosuppressive effects.
Vaccinations
Patients without a healthcare professional-confirmed history of chickenpox or without documentation of a full course of vaccination against VZV should be tested for antibodies to VZV before initiating ZEPOSIA. A full course of vaccination for antibody-negative patients with varicella vaccine is recommended prior to commencing treatment with ZEPOSIA, following which initiation of treatment with ZEPOSIA should be postponed for 4 weeks to allow the full effect of vaccination to occur.
No clinical data are available on the efficacy and safety of vaccinations in patients taking ZEPOSIA. Vaccinations may be less effective if administered during ZEPOSIA treatment.
If live attenuated vaccine immunizations are required, administer at least 1 month prior to initiation of ZEPOSIA. Avoid the use of live attenuated vaccines during and for 3 months after treatment with ZEPOSIA.
5.2 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically occurs in patients who are immunocompromised, and that usually leads to death or severe disability. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
PML has been reported in patients treated with S1P receptor modulators, including ZEPOSIA, and other multiple sclerosis (MS) and UC therapies and has been associated with some risk factors (e.g., immunocompromised patients, polytherapy with immunosuppressants). Physicians should be vigilant for clinical symptoms or MRI findings that may be suggestive of PML. MRI findings may be apparent before clinical signs or symptoms. If PML is suspected, treatment with ZEPOSIA should be suspended until PML has been excluded by an appropriate diagnostic evaluation.
If PML is confirmed, treatment with ZEPOSIA should be discontinued.
Immune reconstitution inflammatory syndrome (IRIS) has been reported in MS patients treated with S1P receptor modulators who developed PML and subsequently discontinued treatment. IRIS presents as a clinical decline in the patient’s condition that may be rapid, can lead to serious neurological complications or death, and is often associated with characteristic changes on MRI. The time to onset of IRIS in patients with PML was generally within a few months after S1P receptor modulator discontinuation. Monitoring for development of IRIS and appropriate treatment of the associated inflammation should be undertaken.
5.3 Bradyarrhythmia and Atrioventricular Conduction Delays
Since initiation of ZEPOSIA may result in a transient decrease in heart rate and atrioventricular conduction delays, an up-titration scheme should be used to reach the maintenance dosage of ZEPOSIA [see Dosage and Administration (2.2) and Clinical Pharmacology (12.2)].
ZEPOSIA was not studied in patients who had:
• A myocardial infarction, unstable angina, stroke, TIA, or decompensated heart failure requiring hospitalization within the last 6 months• New York Heart Association Class III / IV heart failure• Cardiac conduction or rhythm disorders, including sick sinus syndrome, significant QT prolongation (QTcF > 450 msec in males, > 470 msec in females), risk factors for QT prolongation, or other conduction abnormalities or cardiac condition that in the opinion of the treating investigator could jeopardize the patient's health• Other pre-existing stable cardiac conditions without clearance from a cardiologist• Severe untreated sleep apnea• A resting heart rate less than 55 beats per minute (bpm) at baseline
Reduction in Heart Rate
Initiation of ZEPOSIA may result in a transient decrease in heart rate. After the initial dose of ZEPOSIA 0.23 mg, the greatest mean decrease from baseline in heart rate occurred at Hour 5 on Day 1 (decrease of 1.2 bpm in MS Study 1 and Study 2, and 0.7 bpm in UC Study 1 and Study 3), returning to near baseline at Hour 6. With continued up-titration, the maximal heart rate effect of ozanimod occurred on Day 8. The utility of performing first-dose cardiac monitoring when initiating ZEPOSIA in patients with characteristics similar to those studied in the clinical trials of ZEPOSIA is unclear. Heart rates below 40 bpm were not observed. Initiation of ZEPOSIA without titration may result in greater decreases in heart rate [see Dosage and Administration (2.2)].
In MS Study 1 and Study 2, bradycardia was reported on the day of treatment initiation in 0.6% of patients treated with ZEPOSIA compared to no patients who received IFN beta-1a. After Day 1, the incidence of bradycardia was 0.8% in patients treated with ZEPOSIA compared to 0.7% of patients who received IFN beta-1a. In UC Study 1 and Study 3, bradycardia was reported on the day of treatment initiation in 1 patient (0.2%) treated with ZEPOSIA compared to none in patients who received placebo. After Day 1, bradycardia was reported in 1 patient (0.2%) treated with ZEPOSIA. In UC Study 2, bradycardia was not reported.
Atrioventricular Conduction Delays
Initiation of ZEPOSIA may result in transient atrioventricular conduction delays. At ZEPOSIA exposures higher than the recommended dosage without dose titration, first- and second-degree type 1 atrioventricular blocks were observed in healthy volunteers; however, in MS Study 1 and Study 2 and UC Study 1 and Study 3 with dose titration, Mobitz type 2 second- or third-degree atrioventricular blocks were not reported in patients treated with ZEPOSIA.
If treatment with ZEPOSIA is considered, advice from a cardiologist should be sought for those individuals:
• With significant QT prolongation (QTcF > 450 msec in males, > 470 msec in females)• With arrhythmias requiring treatment with Class Ia or Class III anti-arrhythmic drugs• With ischemic heart disease, heart failure, history of cardiac arrest or myocardial infarction, cerebrovascular disease, and uncontrolled hypertension• With a history of with second-degree Mobitz type II or higher AV block, sick-sinus syndrome, or sinoatrial heart block [see Contraindications (4)]5.4 Liver Injury
Elevations of aminotransferases may occur in patients receiving ZEPOSIA.
Obtain transaminase and bilirubin levels, if not recently available (i.e., within 6 months), before initiation of ZEPOSIA.
In MS Study 1 and Study 2, elevations of ALT to 5-fold the upper limit of normal (ULN) or greater occurred in 1.6% of patients treated with ZEPOSIA 0.92 mg and 1.3% of patients who received IFN beta-1a. Elevations of 3-fold the ULN or greater occurred in 5.5% of patients treated with ZEPOSIA and 3.1% of patients who received IFN beta-1a. The median time to an elevation of 3-fold the ULN was 6 months. The majority (79%) of patients continued treatment with ZEPOSIA with values returning to less than 3 times the ULN within approximately 2-4 weeks. ZEPOSIA was discontinued for a confirmed elevation greater than 5-fold the ULN. Overall, the discontinuation rate because of elevations in hepatic enzymes was 1.1% of patients with MS treated with ZEPOSIA 0.92 mg and 0.8% of patients who received IFN beta-1a.
In UC Study 1, elevations of ALT to 5-fold the ULN or greater occurred in 0.9% of patients treated with ZEPOSIA 0.92 mg and 0.5% of patients who received placebo, and in UC Study 2 elevations occurred in 0.9% of patients and no patients, respectively. In UC Study 1, elevations of ALT to 3-fold the ULN or greater occurred in 2.6% of UC patients treated with ZEPOSIA 0.92 mg and 0.5% of patients who received placebo, and in UC Study 2 elevations occurred in 2.3% of patients and no patients, respectively. In controlled and uncontrolled UC studies, the majority (96%) of patients with ALT greater than 3-fold the ULN continued treatment with ZEPOSIA with values returning to less than 3-fold the ULN within approximately 2 to 4 weeks. Overall, the discontinuation rate because of elevations in hepatic enzymes was 0.4% in patients treated with ZEPOSIA 0.92 mg, and none in patients who received placebo in the controlled UC studies.
Individuals with an AST or ALT greater than 1.5 times ULN were excluded from MS Study 1 and Study 2 and greater than 2 times the ULN for UC Study 1 and Study 3. There are no data to establish that patients with preexisting liver disease are at increased risk to develop elevated liver function test values when taking ZEPOSIA. Dosage adjustment in patients with mild or moderate hepatic impairment (Child-Pugh class A or B) is required [see Dosage and Administration (2.3)], and use of ZEPOSIA in patients with severe hepatic impairment (Child-Pugh class C) is not recommended [see Use in Specific Populations (8.6)].
Patients who develop symptoms suggestive of hepatic dysfunction, such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine, should have hepatic enzymes checked, and ZEPOSIA should be discontinued if significant liver injury is confirmed.
5.5 Fetal Risk
There are no adequate and well-controlled studies in pregnant women. Based on animal studies, ZEPOSIA may cause fetal harm [see Use in Specific Populations (8.1)]. Because it takes approximately 3 months to eliminate ZEPOSIA from the body, women of childbearing potential should use effective contraception to avoid pregnancy during treatment and for 3 months after stopping ZEPOSIA [see Use in Specific Populations (8.3)].
5.6 Increased Blood Pressure
In MS Study 1 and Study 2, patients treated with ZEPOSIA had an average increase of approximately 1 to 2 mm Hg in systolic pressure over patients who received IFN beta-1a, and no effect on diastolic pressure. The increase in systolic pressure was first detected after approximately 3 months of treatment and persisted throughout treatment. Hypertension was reported as an adverse reaction in 3.9% of patients treated with ZEPOSIA 0.92 mg and in 2.1% of patients who received IFN beta-1a. Two patients treated with ZEPOSIA in MS Study 1 and one patient treated with interferon (IFN) beta-1a in Study 2 experienced a hypertensive crisis that was not clearly influenced by a concomitant medication.
The mean increase in systolic blood pressure (SBP) and diastolic blood pressure (DBP) in UC patients treated with ZEPOSIA is similar to patients with MS. In UC Study 1 and Study 3, the average increase from baseline in SBP was 3.7 mm Hg in patients treated with ZEPOSIA and 2.3 mm Hg in patients treated with placebo. In UC Study 2, the average increase from baseline in SBP was 5.1 mm Hg in patients treated with ZEPOSIA and 1.5 mm Hg in patients treated with placebo. There was no effect on DBP.
Hypertension was reported as an adverse reaction in 1.2% of patients treated with ZEPOSIA 0.92 mg and none in patients treated with placebo in UC Study 1 and Study 3, and in 2.2% and 2.2% of patients in UC Study 2, respectively. Hypertensive crisis was reported in two patients receiving ZEPOSIA and one patient receiving placebo.
Blood pressure should be monitored during treatment with ZEPOSIA and managed appropriately.
Certain foods that may contain very high amounts (i.e., more than 150 mg) of tyramine could cause severe hypertension because of potential tyramine interaction in patients taking ZEPOSIA, even at the recommended doses. Because of an increased sensitivity to tyramine, patients should be advised to avoid foods containing a very large amount of tyramine while taking ZEPOSIA.
5.7 Respiratory Effects
Dose-dependent reductions in absolute forced expiratory volume over 1 second (FEV1) were observed in MS patients treated with ZEPOSIA as early as 3 months after treatment initiation. In the MS pooled analyses of Study 1 and Study 2, the decline in absolute FEV1 from baseline in patients treated with ZEPOSIA compared to patients who received IFN beta-1a was 60 mL (95% CI: -100, -20) at 12 months. The mean difference in percent predicted FEV1 at 12 months between patients treated with ZEPOSIA and patients who received IFN beta-1a was 1.9% (95% CI: -2.9, -0.8). Dose-dependent reductions in forced vital capacity (FVC) (absolute value and %-predicted) were also seen at Month 3 in pooled analyses comparing patients treated with ZEPOSIA to patients who received IFN beta-1a [60 mL, 95% CI (-110, -10); 1.4%, 95% CI: (-2.6, -0.2)], though significant reductions were not seen at other timepoints. There is insufficient information to determine the reversibility of the decrease in FEV1 or FVC after drug discontinuation. One patient in MS Study 1 discontinued ZEPOSIA because of dyspnea.
In UC Study 1 the mean difference in decline in absolute FEV1 from baseline in patients treated with ZEPOSIA compared to patients who received placebo was 22 mL (95% CI: -84, 39) at 10 weeks. The mean difference in percent predicted normal (PPN) FEV1 at 10 weeks between patients treated with ZEPOSIA compared to those who received placebo was 0.8% (95% CI: -2.6, 1.0). The difference in reductions in FVC (absolute value and %-predicted) seen at Week 10 in UC Study 1, comparing patients who were treated with ZEPOSIA to those who received placebo was 44 mL, 95% CI (-114, 26); 0.5%, 95% CI (-2.3, 1.2), respectively. There is insufficient information to determine the reversibility of observed decreases in FEV1 or FVC after discontinuation of ZEPOSIA, or whether changes could be progressive with continued use.
Spirometric evaluation of respiratory function should be performed during therapy with ZEPOSIA, if clinically indicated.
5.8 Macular Edema
Sphingosine 1-phosphate (S1P) receptor modulators, including ZEPOSIA, have been associated with an increased risk of macular edema.
In MS Study 1 and Study 2, macular edema was observed in 0.3% of patients treated with ZEPOSIA and in 0.3% of patients who received IFN beta-1a. Macular edema was reported in a total of 1 (0.2%) patient in UC Study 1 and Study 3, and in 1 (0.4%) patient in UC Study 2 treated with ZEPOSIA, and in no patients who received placebo.
An ophthalmic evaluation of the fundus, including the macula, is recommended in all patients at any time if there is any change in vision while taking ZEPOSIA.
Continuation of ZEPOSIA therapy in patients with macular edema has not been evaluated. A decision on whether or not ZEPOSIA should be discontinued needs to take into account the potential benefits and risks for the individual patient.
Macular Edema in Patients with a History of Uveitis or Diabetes Mellitus
Patients with a history of uveitis and patients with a history of diabetes mellitus are at increased risk of macular edema during ZEPOSIA therapy. The incidence of macular edema is also increased in patients with a history of uveitis. In addition to the examination of the fundus, including the macula, prior to treatment, patients with diabetes mellitus or a history of uveitis should have regular follow-up examinations.
5.9 Posterior Reversible Encephalopathy Syndrome
Rare cases of posterior reversible encephalopathy syndrome (PRES) have been reported in patients receiving a S1P receptor modulator. In MS controlled clinical trials with ZEPOSIA, one case of PRES was reported. Should a ZEPOSIA-treated patient develop any unexpected neurological or psychiatric symptoms/signs (e.g., cognitive deficits, behavioral changes, cortical visual disturbances, or any other neurological cortical symptoms/signs), any symptom/sign suggestive of an increase of intracranial pressure, or accelerated neurological deterioration, the physician should promptly schedule a complete physical and neurological examination and should consider an MRI. Symptoms of PRES are usually reversible but may evolve into ischemic stroke or cerebral hemorrhage. Delay in diagnosis and treatment may lead to permanent neurological sequelae. If PRES is suspected, treatment with ZEPOSIA should be discontinued.
5.10 Unintended Additive Immunosuppressive Effects from Prior Treatment with Immunosuppressive or Immune-Modulating Drugs
When switching from drugs with prolonged immune effects, the half-life and mode of action of these drugs must be considered to avoid unintended additive immunosuppressive effects while at the same time minimizing risk of disease reactivation, when initiating ZEPOSIA.
Initiating treatment with ZEPOSIA after treatment with alemtuzumab is not recommended [see Drug Interactions (7)].
5.11 Severe Increase in Multiple Sclerosis Disability after Stopping ZEPOSIA
In MS, severe exacerbation of disease, including disease rebound, has been rarely reported after discontinuation of a S1P receptor modulator. The possibility of severe exacerbation of disease should be considered after stopping ZEPOSIA treatment. Patients should be observed for a severe increase in disability upon ZEPOSIA discontinuation and appropriate treatment should be instituted, as required.
After stopping ZEPOSIA in the setting of PML, monitor for development of immune reconstitution inflammatory syndrome (PML-IRIS) [see Warnings and Precautions (5.2) ].
5.12 Immune System Effects after Stopping ZEPOSIA
After discontinuing ZEPOSIA, the median time for peripheral blood lymphocytes to return to the normal range was approximately 30 days, with approximately 80% to 90% of patients in the normal range within 3 months [see Clinical Pharmacology (12.2)]. Use of immunosuppressants within this period may lead to an additive effect on the immune system, and therefore caution should be applied when initiating other drugs 4 weeks after the last dose of ZEPOSIA [see Drug Interactions (7)].
6 Adverse Reactions
The following serious adverse reactions are described elsewhere in the labeling:
• Infections [see Warnings and Precautions (5.1)]• Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.2)]• Bradyarrhythmia and Atrioventricular Conduction Delays [see Warnings and Precautions (5.3)]• Liver Injury [see Warnings and Precautions (5.4) ]• Fetal Risk [see Warnings and Precautions (5.5) ]• Increased Blood Pressure [see Warnings and Precautions (5.6) ]• Respiratory Effects [see Warnings and Precautions (5.7) ]• Macular Edema [see Warnings and Precautions (5.8) ]• Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.9) ]• Unintended Additive Immunosuppressive Effects from Prior Treatment with Immunosuppressive or Immune-Modulating Drugs [see Warnings and Precautions (5.10) ]• Severe Increase in Multiple Sclerosis Disability after Stopping ZEPOSIA [see Warnings and Precautions (5.11) ]• Immune System Effects after Stopping ZEPOSIA [see Warnings and Precautions (5.12) ]
Most common adverse reactions (incidence ≥4%) are:
• Multiple Sclerosis: upper respiratory infection, hepatic transaminase elevation, orthostatic hypotension, urinary tract infection, back pain, and hypertension. (6.1 )• Ulcerative Colitis: liver test increased, upper respiratory infection, and headache. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Common Adverse Reactions
Multiple Sclerosis
The safety of ZEPOSIA was evaluated in two randomized, double-blind, active comparator-controlled clinical studies in which 882 patients received ZEPOSIA 0.92 mg [see Clinical Studies (14.1)].
Table 2 uls adverse reactions that occurred in at least 2% of ZEPOSIA-treated patients and greater than comparator. The most common adverse reactions that occurred in at least 4% of ZEPOSIA-treated patients and greater than in patients who received IFN beta-1a were upper respiratory infection, hepatic transaminase elevation, orthostatic hypotension, urinary tract infection, back pain, and hypertension.
Table 2: Adverse Reactions with an Incidence of at Least 2% in ZEPOSIA-Treated Patients and at Least 1% Greater than IFN beta-1a in Patients with Multiple Sclerosis (Pooled MS Study 1 and Study 2)a a Data are not an adequate basis for comparison of rates between ZEPOSIA and the active control. b Includes the following terms: nasopharyngitis, upper respiratory tract infection, pharyngitis, respiratory tract infection, bronchitis, rhinitis, viral respiratory tract infection, viral upper respiratory tract infection, rhinorrhea, tracheitis, and laryngitis. c Includes the following terms: alanine aminotransferase increased, gamma-glutamyl transferase increased, aspartate aminotransferase increased, hepatic enzyme increased, abnormal liver function test, and increased transaminases. d Includes hypertension, essential hypertension, and orthostatic hypertension. e ZEPOSIA was initiated with a 7-day titration [see Dosage and Administration (2.2)].
Adverse Reactions
MS Studies 1 and 2
ZEPOSIA 0.92 mg Once Dailye (n=882) %
IFN beta-1a 30 mcg Intramuscularly Once Weekly (n=885) %
Upper respiratory infectionb
26
23
Hepatic transaminase elevationc
10
5
Orthostatic hypotension
4
3
Urinary tract infection
4
3
Back pain
4
3
Hypertensiond
4
2
Upper abdominal pain
2
1
Ulcerative Colitis
The safety of ZEPOSIA was evaluated in two randomized, double-blind, placebo-controlled clinical studies [UC Study 1 (induction), n=429; and UC Study 2 (maintenance), n=230] in adult patients with moderately to severely active ulcerative colitis [see Clinical Studies (14.2)]. Additional data from the induction period of a randomized, double-blind, placebo-controlled study (UC Study 3, NCT01647516) included 67 patients who received ZEPOSIA 0.92 mg once daily.
Common adverse reactions in UC Study 1 and Study 3 and in UC Study 2 are uled in Tables 3 and 4, respectively. The most common adverse reactions that occurred in at least 4% of ZEPOSIA-treated patients and greater than in patients who received placebo were liver test increased, upper respiratory infection, and headache.
Table 3: Adverse Reactions with an Incidence of at Least 2% in ZEPOSIA-Treated Patients and at Least 1% Greater than Placebo in Patients with Ulcerative Colitis (Pooled UC Study 1 and Study 3) a Includes the following terms: streptococcal pharyngitis, pharyngotonsillitis, bacterial pharyngitis, nasopharyngitis, upper respiratory tract infection, pharyngitis, sinusitis, tonsillitis, viral upper respiratory tract infection, laryngitis, acute sinusitis, catarrh, chronic sinusitis, upper respiratory tract inflammation, chronic tonsillitis, viral pharyngitis, viral sinusitis, bacterial sinusitis, bacterial upper respiratory tract infection, viral labyrinthitis, laryngeal inflammation, and pharyngeal inflammation. b Includes the following terms: gamma-glutamyl transferase increased, alanine aminotransferase increased, aspartate aminotransferase increased, hepatic enzyme increased, hyperbilirubinemia, liver function test increased, blood alkaline phosphatase increased, and increased transaminases. c ZEPOSIA was initiated with a 7-day titration [see Dosage and Administration (2.2)]. d Percentages were calculated as the sum of each individual study percentage multiplied by its Cochran-Mantel-Haenszel weight.
Adverse Reactions
Induction Periods (UC Study 1 and Study 3)
ZEPOSIA 0.92 mg Once Daily (n=496)c,d %
Placebo (n=281) %d
Upper respiratory infectiona
5
4
Liver test increasedb
5
0
Headache
4
3
Pyrexia
3
2
Nausea
3
2
Arthralgia
3
1
Table 4: Adverse Reactions with an Incidence of at Least 4% in ZEPOSIA-Treated Patients and at Least 1% Greater than Placebo in Patients with Ulcerative Colitis (UC Study 2) a Includes the following terms: gamma-glutamyl transferase increased, alanine aminotransferase increased, aspartate aminotransferase increased, hepatic enzyme increased, hyperbilirubinemia, blood bilirubin increased, liver function test increased, and blood alkaline phosphatase increased.
Adverse Reactions
Maintenance Period (UC Study 2)
ZEPOSIA 0.92 mg Once Daily (n=230) %
Placebo (n=227) %
Liver test increaseda
11
2
Headache
5
<1
Other Adverse Reactions
Reduction in Heart Rate
Initiation of ZEPOSIA may result in transient decrease in heart rate in MS and UC patients [see Warnings and Precautions (5.3)].
Respiratory Effects
Dose-dependent reductions in absolute FEV1 and FVC were observed in MS and UC patients treated with ZEPOSIA [see Warnings and Precautions (5.7) ].
Malignancies
Malignancies, such as melanoma, basal cell carcinoma, breast cancer, seminoma, cervical carcinoma, and adenocarcinomas, including rectal adenocarcinoma, were reported with ZEPOSIA in controlled trials of MS and UC. An increased risk of cutaneous malignancies has been reported with another S1P receptor modulator.
Hypersensitivity
Hypersensitivity, including rash and urticaria, has been reported with ZEPOSIA in active-controlled MS clinical trials.
Peripheral Edema
Peripheral edema was observed in 3% of ZEPOSIA-treated patients and in 0.4% of patients who received placebo in UC Study 2.
7 Drug Interactions
Tables 5 and 6 include drugs with clinically important drug, tyramine, and vaccine interactions when administered concomitantly with ZEPOSIA and instructions for preventing or managing them.
Table 5: Clinically Relevant Interactions Affecting Drugs, Tyramine, and Vaccines Co-administered with ZEPOSIA
Anti-Neoplastic, Immune-Modulating, or Non-Corticosteroid Immunosuppressive Therapies
Clinical Impact:
ZEPOSIA has not been studied in combination with anti-neoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies with the exception of cyclosporine, which had no pharmacokinetic interaction [see Clinical Pharmacology (12.3)].
Prevention or Management:
Caution should be used during concomitant administration because of the risk of additive immune effects during such therapy and in the weeks following administration [see Warnings and Precautions (5.1)].When switching from drugs with prolonged immune effects, the half-life and mode of action of these drugs must be considered in order to avoid unintended additive immunosuppressive effects [see Warnings and Precautions (5.10) ]. Alemtuzumab: Initiating treatment with ZEPOSIA after alemtuzumab is not recommended because of the characteristics and duration of alemtuzumab immune suppressive effects. Beta interferon or glatiramer acetate: ZEPOSIA can generally be started immediately after discontinuation of beta interferon or glatiramer acetate.
Anti-Arrhythmic Drugs, QT Prolonging Drugs, Drugs That May Decrease Heart Rate
Clinical Impact:
ZEPOSIA has not been studied in patients taking QT prolonging drugs.Class Ia (e.g., quinidine, procainamide) and Class III (e.g., amiodarone, sotalol) anti-arrhythmic drugs have been associated with cases of Torsades de Pointes in patients with bradycardia.
Prevention or Management:
If treatment with ZEPOSIA is considered in patients on Class Ia or Class III anti-arrhythmic drugs, advice from a cardiologist should be sought [see Warnings and Precautions (5.3)].Because of the potential additive effects on heart rate, treatment with ZEPOSIA should generally not be initiated in patients who are concurrently treated with QT prolonging drugs with known arrhythmogenic properties [see Warnings and Precautions (5.3)]. If treatment initiation with ZEPOSIA is considered in patients on QT prolonging drugs, advice from a cardiologist should be sought.
Adrenergic and Serotonergic Drugs
Clinical Impact:
Because an active metabolite of ozanimod inhibits MAO-B in vitro, there is a potential for serious adverse reactions, including hypertensive crisis with co-administration of ZEPOSIA with drugs or over-the-counter medications that can increase norepinephrine or serotonin [e.g., opioid drugs, selective serotonin reuptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors (SNRIs), tricyclics, tyramine]. Opioid Drugs Serious, sometimes fatal reactions have been precipitated with concomitant use of opioid drugs (e.g., meperidine and its derivatives, methadone, or tramadol) and MAOIs, including selective MAO-B inhibitors. Although a small number of patients treated with ZEPOSIA were concomitantly exposed to opioids, this exposure was not adequate to rule out the possibility of an adverse reaction from co-administration. Serotonergic Drugs Although a small number of patients treated with ZEPOSIA were concomitantly exposed to serotonergic medications, this exposure was not adequate to rule out the possibility of an adverse reaction from co-administration. Sympathomimetic Medications Concomitant use of ZEPOSIA with pseudoephedrine did not potentiate the effects on blood pressure [see Clinical Pharmacology (12.2)]. However, hypertensive crisis has occurred with administration of ZEPOSIA alone [see Warnings and Precautions (5.6) ] and hypertensive crisis has been reported with co-administration of other selective and nonselective MAO inhibitors (e.g., rasagiline) with sympathomimetic medications.
Prevention or Management:
Co-administration of ZEPOSIA with drugs or over-the-counter medications that can increase norepinephrine or serotonin (e.g., opioid drugs, SSRIs, SNRIs, tricyclics, tyramine) is not recommended. Monitor patients for hypertension with concomitant use.
Combination Beta Blocker and Calcium Channel Blocker
Clinical Impact:
The co-administration of ZEPOSIA with both a beta blocker and a calcium channel blocker has not been studied. However, there is a potential of additive effects on heart rate.
Prevention or Management:
Treatment with ZEPOSIA should generally not be initiated in patients who are concurrently treated with both a heart rate lowering calcium channel blocker (e.g., verapamil, diltiazem) and beta blocker [see Warnings and Precautions (5.3)]. If treatment initiation with ZEPOSIA is considered in patients on both a heart rate lowering calcium channel blocker and beta blocker, advice from a cardiologist should be sought.
Tyramine
Clinical Impact:
MAO in the gastrointestinal tract and liver (primarily type A) provides protection from exogenous amines (e.g., tyramine). If tyramine were absorbed intact, it could lead to severe hypertension, including hypertensive crisis. Aged, fermented, cured, smoked, and pickled foods containing large amounts of exogenous amines (e.g., aged cheese, pickled herring) may cause release of norepinephrine resulting in a rise in blood pressure (tyramine reaction).
Prevention or Management:
Patients should be advised to avoid foods containing a large amount of tyramine while taking recommended doses of ZEPOSIA [see Warnings and Precautions (5.6) ].
Vaccination
Clinical Impact:
During, and for up to three months after, discontinuation of treatment with ZEPOSIA, vaccinations may be less effective. The use of live attenuated vaccines may carry the risk of infection.
Prevention or Management:
Live attenuated vaccines should be avoided during ZEPOSIA treatment and for up to 3 months after discontinuation of treatment with ZEPOSIA [see Warnings and Precautions (5.1)].
Table 6: Clinically Relevant Interactions Affecting ZEPOSIA When Co-administered with Other Drugs
Monoamine Oxidase (MAO) Inhibitors
Clinical Impact:
Co-administration of ZEPOSIA with MAO-B inhibitors may decrease exposure of the active metabolites of ozanimod. In addition, metabolites of ozanimod may inhibit MAO [see Clinical Pharmacology (12.3)]. The potential for a clinical interaction with MAO inhibitors has not been studied; however, the increased risk of nonselective MAO inhibition may lead to a hypertensive crisis.
Prevention or Management:
Co-administration of ZEPOSIA with MAO inhibitors (e.g., selegiline, phenelzine, linezolid) is contraindicated. At least 14 days should elapse between discontinuation of ZEPOSIA and initiation of treatment with MAO inhibitors.
Strong CYP2C8 Inhibitors
Clinical Impact:
Co-administration of ZEPOSIA with strong CYP2C8 inhibitors increases the exposure of the active metabolites of ozanimod [see Clinical Pharmacology (12.3)], which may increase the risk of ZEPOSIA adverse reactions.
Prevention or Management:
Co-administration of ZEPOSIA with strong CYP2C8 inhibitors (e.g., gemfibrozil) is not recommended.
Strong CYP2C8 Inducers
Clinical Impact:
Co-administration of ZEPOSIA with strong CYP2C8 inducers (e.g., rifampin) reduces the exposure of the major active metabolites of ozanimod [see Clinical Pharmacology (12.3)], which may decrease the efficacy of ZEPOSIA.
Prevention or Management:
Co-administration of ZEPOSIA with strong CYP2C8 inducers should be avoided.
• Vaccination: Avoid use of live attenuated vaccines during and for up to 3 months after treatment with ZEPOSIA. (7 )• See full prescribing information for a ul of clinically important drug interactions. (7 )
8 Use In Specific Populations
Hepatic Impairment: Use in severe hepatic impairment (Child-Pugh class C) is not recommended. (8.6 )
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ZEPOSIA during pregnancy. Healthcare providers are encouraged to register patients on-line, or pregnant women may register themselves at https://www.zeposiapregnancyregistry.com/or by calling 1-877-301-9314. Currently this registry is enrolling women with MS. Information regarding registration of pregnant women with UC will be made available in the future.
Risk Summary
There are no adequate data on the developmental risk associated with the use of ZEPOSIA in pregnant women. In animal studies, administration of ozanimod during pregnancy produced adverse effects on development, including embryolethality, an increase in fetal malformations, and neurobehavioral changes, in the absence of maternal toxicity. In rabbits, fetal blood vessel malformations occurred at clinically relevant maternal ozanimod and metabolite exposures (see Data). The receptor affected by ozanimod (sphingosine1-phosphate) has been demonstrated to have an important role in embryogenesis, including vascular and neural development.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of ozanimod (0, 0.2, 1, or 5 mg/kg/day) to female rats during organogenesis resulted in a marked increase in embryofetal mortality, increased fetal malformations and skeletal variations (abnormal/delayed ossification), and reduced fetal body weight at the highest dose tested. No maternal toxicity was observed. At the no-effect dose (1 mg/kg/day) for adverse effects on embryofetal development, plasma ozanimod exposure (AUC) for ozanimod was approximately 60 times that in humans at the maximum recommended human dose (MRHD) of 0.92 mg/day. Plasma AUCs for major human metabolites, CC112273 and CC1084037, were similar to and less than, respectively, those in humans at the MRHD.
Oral administration of ozanimod (0, 0.2, 0.6, or 2.0 mg/kg/day) to female rabbits during organogenesis resulted in a marked increase in embryofetal mortality at the highest dose tested and increased fetal malformations (malformed blood vessels) and skeletal variations at the mid and high doses. Maternal toxicity was not observed. At the no-effect dose (0.2 mg/kg/day) for adverse effects on embryofetal development in rabbit, plasma ozanimod exposure (AUC) was approximately 2 times that in humans at the MRHD; plasma AUCs for major human metabolites, CC112273 and CC1084037, were less than those in humans at the MRHD.
Oral administration of ozanimod (0, 0.2, 0.7, or 2 mg/kg/day) to female rats throughout gestation and lactation resulted in persistent body weight reductions and long-term effects on reproductive (prolonged estrus cycle) and neurobehavioral (increased motor activity) function in offspring at the highest dose tested, which was not associated with maternal toxicity. At the no-effect dose (0.7 mg/kg/day) for adverse effects on pre- and postnatal development, plasma ozanimod exposure (AUC) was 30 times that in humans at the MRHD; plasma AUCs for major human metabolites, CC112273 and CC1084037, were less than those in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of ozanimod in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Following oral administration of ozanimod, ozanimod and/or metabolites were detected in the milk of lactating rat at levels higher than those in maternal plasma.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZEPOSIA and any potential adverse effects on the breastfed infant from ZEPOSIA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Before initiation of ZEPOSIA treatment, women of childbearing potential should be counseled on the potential for a serious risk to the fetus and the need for contraception during treatment with ZEPOSIA [see Use in Specific Populations (8.1)]. Because of the time it takes to eliminate the drug from the body after stopping treatment, the potential risk to the fetus may persist and women of childbearing age should also use effective contraception for 3 months after stopping ZEPOSIA.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of ZEPOSIA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. No clinically significant differences in the pharmacokinetics of ozanimod and CC112273 were observed based on age [see Clinical Pharmacology (12.3)]. Monitor elderly patients for cardiac and hepatic adverse reactions, because of the greater frequency of reduced cardiac and hepatic function in the elderly population.
8.6 Hepatic Impairment
In patients with mild (Child-Pugh class A) or moderate hepatic impairment (Child-Pugh class B), the exposures of ozanimod and its active metabolites are higher than those in healthy controls [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions. Therefore, dosage adjustment in patients with mild or moderate hepatic impairment is required [see Dosage and Administration (2.3)].
The pharmacokinetics of ozanimod and its active metabolites were not evaluated in patients with severe hepatic impairment (Child-Pugh class C). Therefore, use of ZEPOSIA in patients with severe hepatic impairment is not recommended.
11 Description
ZEPOSIA contains ozanimod, a sphingosine 1-phosphate receptor modulator and is supplied as ozanimod hydrochloride (HCl).
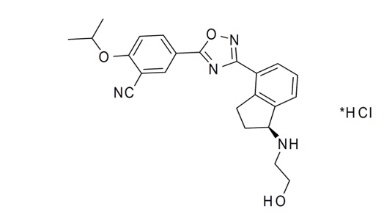
The chemical name of ozanimod HCl is 5-(3-{(1S)-1-[(2-hydroxyethyl)amino]-2,3-dihydro-1H-inden-4-yl}-1,2,4-oxadiazol-5-yl)-2-[(propan-2-yl)oxy]benzonitrile, monohydrochloride.
Ozanimod HCl is a white to off-white solid that is freely soluble in water and alcohol with a molecular weight of 440.92 g/mol.
The chemical structure is:
ZEPOSIA capsules are provided as hard gelatin capsules for oral administration, containing 0.23, 0.46, or 0.92 mg of ozanimod (equivalent to 0.25, 0.5, and 1 mg ozanimod HCl, respectively). ZEPOSIA capsules consist of the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The capsule shell, imprinted with black ink, contains the following inactive ingredients: black iron oxide, gelatin, red iron oxide, titanium dioxide, and yellow iron oxide.
12 Clinical Pharmacology
12.1 Mechanism of Action
Ozanimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. Ozanimod blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. Ozanimod has minimal or no activity on S1P2, S1P3, and S1P4. The mechanism by which ozanimod exerts therapeutic effects in multiple sclerosis and ulcerative colitis is unknown but may involve the reduction of lymphocyte migration into the central nervous system and intestine.
12.2 Pharmacodynamics
Reduction in Blood Lymphocyte Counts
In active-controlled MS and controlled UC clinical trials, mean lymphocyte counts decreased to approximately 45% of baseline at 3 months (approximate mean blood lymphocyte counts 0.8 Ă— 109/L), and low lymphocyte counts were maintained during treatment with ZEPOSIA [see Warnings and Precautions (5.1)].
After discontinuing ZEPOSIA 0.92 mg, the median time for peripheral blood lymphocytes to return to the normal range was 30 days, with approximately 90% of patients in the normal range within 3 months.
Reduction in Heart Rate
ZEPOSIA may cause a transient decrease in heart rate on initiation of dosing [see Warnings and Precautions (5.3)]. An up-titration schedule of ZEPOSIA 0.23 mg followed by doses of 0.46 mg, and 0.92 mg attenuates the magnitude of heart rate reductions [see Dosage and Administration (2.2)].
Drug Interaction Studies
Sympathomimetic Agents
No clinically significant differences in heart rate or blood pressure was observed when ZEPOSIA 1.84 mg daily (two times the recommended dosage) for 28 days was co-administered with a single dose of 60 mg pseudoephedrine (a sympathomimetic agent) compared to pseudoephedrine alone [see Drug Interactions (7)].
Beta Blocker or Calcium Channel Blocker
The effect of co-administration of the maintenance dosage of ZEPOSIA, propranolol, or diltiazem, or administration with both a beta blocker and a calcium channel blocker taken together has not been studied [see Drug Interactions (7)].
Pulmonary Function
Dose-dependent reductions in FEV1 and FVC were observed in patients treated with ZEPOSIA [see Warnings and Precautions (5.7) ].
Cardiac Electrophysiology
Following a 14-day titration regimen of once daily doses of ozanimod 0.23 mg for 4 days, 0.46 mg for 3 days, 0.92 mg for 3 days, and 1.84 mg (2 times the maximum approved recommended dose) for 4 days in healthy subjects, ZEPOSIA did not prolong the QTc interval to any clinically relevant extent [see Warnings and Precautions (5.3)].
12.3 Pharmacokinetics
The steady state exposure parameters of ozanimod and its major active metabolite, CC112273 are summarized in Table 7. Population pharmacokinetic analysis indicated no meaningful differences in these pharmacokinetic parameters in patients with relapsing MS or UC.
Table 7: Exposure Parameters of Ozanimod and its Major Metabolitea a Mean [coefficient of variation (CV%)] following ozanimod 0.92 mg once daily dose in relapsing MS patients, unless otherwise specified. b In healthy subjects.Cmax,ss = maximum observed plasma concentration at steady state, AUCtau,ss = area under the plasma concentration-time curve during a dosage interval at steady state.
Parameters
Ozanimod
CC112273
Cmax,ss
0.244 ng/mL (31.8%)
6.98 ng/mL (42.7%)
AUCtau,ss
4.46 ng*h/mL (31.8%)
143.77 ng*h/mL (39.2%)
Dose Proportionality
The Cmax and AUC increases proportionally over the ozanimod dose range from 0.46 mg to 0.92 mg.
Time to Steady State
102 hours (28.2%)b
45 days (45%)
Accumulation Ratio
2.40 (21.1%) b
16 (101%)
Absorption
The Tmax of ozanimod is approximately 6 to 8 hours.
Effect of Food
No clinically significant differences in the Cmax and AUC of ozanimod were observed following administration of ZEPOSIA with either a high-fat, high-calorie meal (1000 calories, 50% fat) or a low-fat, low-calorie meal (300 calories, 10% fat) compared to fasted conditions [see Dosage and Administration (2.2)].
Distribution
The mean (CV%) apparent volume of distribution of ozanimod (Vz/F) is 5590 L (27%). Human plasma proteins binding of ozanimod, CC112273 and CC1084037 is approximately 98.2%, 99.8%, and 99.3%, respectively.
Elimination
The mean (CV%) plasma half-life (t1/2) of ozanimod is approximately 21 hours (15%). The mean (CV%) effective half-life (t1/2) of CC112273 and its direct interconverting metabolite CC1084037 was approximately 11 days (104%) in relapsing MS patients. The mean (CV%) apparent oral clearance for ozanimod was approximately 192 L/h (37%).
Metabolism
Ozanimod is metabolized by multiple enzymes to form circulating major active metabolites (e.g., CC112273 and CC1084037) and minor active metabolites (e.g., RP101988, RP101075, and RP112509) with similar activity and selectivity for S1P1 and S1P5. Ozanimod is metabolized by ALDH/ADH to form carboxylate metabolite RP101988 and by CYP3A4 to form RP101075. RP101075 is then metabolized either by NAT-2 to form a minor active metabolite RP101442 or by MAO-B to form CC112273. CC112273 is then metabolized by CYP2C8 to form RP112509 or reduced to form CC1084037. CC1084037 is metabolized by AKR 1C1/1C2 and/or 3β- and 11β-HSD to form CC112273. The interconversion between CC112273 and CC1084037 favors CC112273. Approximately 94% of circulating total active drug exposure is represented by ozanimod (6%), CC112273 (73%), and CC1084037 (15%), in humans.
Excretion
Following a single oral dose of radiolabeled ozanimod 0.92 mg, approximately 26% of the radioactivity was recovered in urine and 37% in feces, primarily composed of inactive metabolites.
Specific Populations
Geriatric Patients
Population pharmacokinetic analyses showed that steady state exposure (AUC) of CC112273 in UC patients over 65 years of age was approximately 3% to 4% greater than patients 45 to 65 years of age and 27% greater than adult patients under 45 years of age. There is no meaningful difference in the pharmacokinetics in elderly patients with UC [see Use in Specific Populations (8.5)].
Male and Female Patients
No clinically significant differences in the pharmacokinetics of ozanimod and CC112273 were observed based on sex or weight.
Racial or Ethnic Groups
In a dedicated Japanese PK bridging study, following repeated dosing of 0.96 mg ZEPOSIA, ozanimod exposures (Cmax and AUCtau) were unchanged and CC112273 exposures (Cmax and AUCtau) were approximately 28% and 43% higher, respectively, in Japanese subjects (N=10) compared to Caucasian subjects (N=12). These differences are not considered clinically meaningful.
Patients with Renal Impairment
In a dedicated renal impairment trial, following a single oral dose of 0.23 mg ZEPOSIA, exposures (AUClast) for ozanimod and CC112273 were approximately 27% higher and 23% lower, respectively, in subjects with end stage renal disease (N=8) compared to subjects with normal renal function (N=8). Based on this trial, renal impairment has no clinically important effects on pharmacokinetics of ozanimod or CC112273.
Patients with Hepatic Impairment
In a hepatic impairment study, following an 8-day titration regimen of once daily doses of ZEPOSIA 0.23 mg on days 1 to 4, 0.46 mg on days 5 to 7, and 0.92 mg on day 8, in subjects with mild hepatic impairment (Child-Pugh class A, N=8), mean exposures (AUClast) of ozanimod and active metabolites CC112273 and CC1084037 on day 8 increased by 60%, 98%, and 107%, respectively, compared to healthy controls (N=8). In subjects with moderate hepatic impairment (Child-Pugh class B, N=8) mean exposures (AUClast) of ozanimod and active metabolites CC112273 and CC1084037 on day 8 increased by 17%, 38%, and 61%, respectively, compared to healthy controls.
The pharmacokinetics of ozanimod or its active metabolites were not evaluated in patients with severe hepatic impairment (Child-Pugh class C) [see Use in Specific Populations (8.6)].
Smokers
Population PK analyses showed that CC112273 steady-state exposure (AUC) was approximately 50% lower in smokers than in nonsmokers, although for smokers this reduction in exposure did not result in meaningful differences in absolute lymphocyte count (ALC) reduction or an apparent impact on clinical efficacy.
Drug Interaction Studies
Clinical Studies
In Vitro Studies
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of ozanimod (0, 8, 25, or 80 mg/kg/day) to Tg.rasH2 mice for 26-weeks resulted in an increase in hemangioma and hemangiosarcoma (combined) in males and females at the mid and high doses tested.
Oral administration of ozanimod (0, 0.2, 0.7, or 2 mg/kg/day) to rats for 2 years resulted in no increase in tumors. At the highest dose tested (2 mg/kg/day), plasma exposure (AUC) for ozanimod was approximately 100 times that in humans at the maximum recommended human dose (MRHD) of 0.92 mg/day. Plasma AUCs for major human metabolites, CC112273 and CC1084037, were similar to and less than, respectively, those in humans at the MRHD.
Mutagenesis
Ozanimod was negative in a battery of in vitro (Ames, mouse lymphoma tk) and in vivo (rat micronucleus) assays. Metabolite CC112273 was negative in in vitro (Ames, chromosomal aberration in mammalian cell) assays. Metabolite CC1084037 was negative in an Ames assay, and positive in an in vitro chromosomal aberration assay in human (TK6) cells but negative in an in vivo rat micronucleus/comet assay.
Impairment of Fertility
Oral administration of ozanimod (0, 0.2, 2, or 30 mg/kg/day) to male and female rats prior to and during mating and continuing through gestation day 7 resulted in no adverse effects on fertility. At the highest dose tested (30 mg/kg/day), plasma ozanimod exposure (AUC) was approximately 1600 times that in humans at the maximum recommended human dose (MRHD) (0.92 mg/day); plasma AUCs for metabolites, CC112273 and CC1084037, at 30 mg/kg/day were 13 and 3 times, respectively, those in humans at the MRHD.
14 Clinical Studies
14.1Multiple Sclerosis
The efficacy of ZEPOSIA was demonstrated in 2 randomized, double-blind, double-dummy, parallel-group, active comparator-controlled clinical trials of similar design, in patients with relapsing forms of MS [Study 1 (NCT02294058) and Study 2 (NCT02047734)]. Patients in Study 1 were treated until the last enrolled patient completed 1 year of treatment. Patients in Study 2 were treated for 24 months. Both studies included patients who had experienced at least 1 relapse within the prior year, or 1 relapse within the prior 2 years with evidence of at least a gadolinium-enhancing (GdE) lesion in the prior year, and had an Expanded Disability Status Scale (EDSS) score from 0 to 5.0 at baseline. Patients with primary progressive MS were excluded.
Patients were randomized to receive either ZEPOSIA 0.92 mg given orally once daily, beginning with a dosage titration [see Dosage and Administration (2.2)], or interferon (IFN) beta-1a, the active comparator, 30 mcg given intramuscularly once weekly. Neurological evaluations were performed at baseline, every 3 months, and at the time of a suspected relapse. Brain MRI scans were performed at baseline, 6 months (Study 1), 1 year (Studies 1 and 2), and 2 years (Study 2).
The primary endpoint of both Study 1 and Study 2 was the annualized relapse rate (ARR) over the treatment period (Study 1) and 24 months (Study 2). Additional outcome measures included: 1) the number of new or enlarging MRI T2 hyperintense lesions over 12 and 24 months, 2) the number of MRI T1 Gadolinium-enhancing (Gd+) lesions at 12 and 24 months, and 3) the time to confirmed disability progression, defined as at least a 1-point increase from baseline EDSS confirmed after 3 months and after 6 months. Confirmed disability progression was evaluated in a pooled analysis of Studies 1 and 2.
In Study 1, a total of 895 patients were randomized to receive ZEPOSIA (n=447) or IFN beta-1a (n=448); of these patients, 94% who received ZEPOSIA and 92% who received IFN beta-1a completed the study. The mean age was 35.4 years, 99.8% were White, and 65% were female. The mean time since MS symptom-onset was 6.9 years, and the median EDSS score at baseline was 2.5; 31% had been treated with a non-steroid therapy for MS. At baseline, the mean number of relapses in the prior year was 1.3 and 48% of patients had one or more T1 Gd-enhancing lesions (mean 1.8) on their baseline MRI scan.
In Study 2, a total of 874 patients were randomized to receive ZEPOSIA (n=433) or IFN beta-1a (n=441); of these patients, 90% who received ZEPOSIA and 85% who received IFN beta-1a completed the study. The mean age was 35.6 years, 98% were White, and 68% were female. The mean time since MS symptom onset was 6.6 years, and the median EDSS score at baseline was 2.5; 29% of patients had been treated with a non-steroid therapy for MS. At baseline, the mean number of relapses in the prior year was 1.3 and 43% of patients had one or more T1 Gd-enhancing lesions (mean 1.7).
The ARR was statistically significantly lower in patients treated with ZEPOSIA 0.92 mg than in patients who received IFN beta-1a 30 mcg IM. The number of new or enlarging T2 lesions and the number of GdE lesions were statistically significantly lower in patients treated with ZEPOSIA 0.92 mg than in patients who received IFN beta-1a.
There was no statistically significant difference in the three-month and six-month confirmed disability progression between ZEPOSIA and IFN beta-1a-treated patients over 2 years.
The results for Study 1 and Study 2 are shown in Table 8.
Table 8: Clinical and MRI Endpoints from MS Study 1 and Study 2 a Through the treatment period (mean duration 13.6 months). b Over treatment period for Study 1 and over 24 months for Study 2. c Disability progression defined as 1-point increase in Expanded Disability Status Scale (EDSS) confirmed 3 months or 6 months later. d Prospective planned pooled analysis of Studies 1 and 2. e Not statistically significant. f Over 12 months for Study 1 and over 24 months for Study 2. g At 12 months for Study 1 and at 24 months for Study 2.
Endpoints
Study 1
Study 2
ZEPOSIA 0.92 mg (n=447) %
IFN beta-1a 30 mcg (n=448) %
ZEPOSIA 0.92 mg (n=433) %
IFN beta-1a 30 mcg (n=441) %
Clinical Endpoints
Annualized Relapse Rate (Primary Endpoint)
0.181a
0.350 a
0.172
0.276
  Relative Reduction
48% (p<0.0001)
38% (p<0.0001)
Percentage of patients without relapseb
78%
66%
76%
64%
Proportion of Patients with 3-Month Confirmed Disability Progressionc,d
7.6% ZEPOSIA vs. 7.8% IFN beta-1a
  Hazard Ratio
0.95 (p=0.77)e
MRI Endpoints
Mean number of new or enlarging T2 hyperintense lesions per MRIf
1.47
2.84
1.84
3.18
  Relative Reduction
48% (p<0.0001)
42% (p<0.0001)
Mean number of T1 Gd-enhancing lesionsg
0.16
0.43
0.18
0.37
  Relative Reduction
63% (p<0.0001)
53% (p=0.0006)
A similar effect of ZEPOSIA on the ARR compared to IFN beta-1a was observed in exploratory subgroups defined by sex, age, prior non-steroid therapy for MS, and baseline disease activity.
14.2Ulcerative Colitis
The efficacy and safety of ZEPOSIA were evaluated in two multicenter, randomized, double-blind, placebo-controlled clinical studies [UC Study 1 (induction) and UC Study 2 (maintenance) (NCT02435992)] in adult patients with moderately to severely active ulcerative colitis.
UC Study 1
In UC Study 1, a total of 645 patients were randomized 2:1 to either ZEPOSIA 0.92 mg given orally once daily or placebo for 10 weeks, beginning with a dosage titration [see Dosage and Administration (2.2)]. The trial included adult patients with moderately to severely active UC who had an inadequate response or were intolerant to any of the following: oral aminosalicylates, corticosteroids, immunomodulators (e.g., 6-mercaptopurine and azathioprine), or a biologic (e.g., TNF blocker and/or vedolizumab). Patients were required to be on stable doses of oral aminosalicylates and/or corticosteroids (prednisone daily dose up to 20 mg equivalent or budesonide extended-release tablets) prior to enrollment. Seventy-one percent of patients were receiving mesalamine, 13% sulfasalazine, and 33% oral corticosteroids. A total of 30% of patients had previously failed or were intolerant to TNF blockers. Of these patients, 63% received at least two biologics including TNF blockers.
The disease activity was assessed by the Mayo score (0 to 12) which consists of four subscores (0 to 3 for each subscore): stool frequency, rectal bleeding, findings on centrally-read endoscopy, and physician global assessment. An endoscopy subscore of 2 was defined by marked erythema, lack of vascular pattern, friability, and erosions; an endoscopy subscore of 3 was defined by spontaneous bleeding and ulceration. Enrolled patients had Mayo scores between 6 to 12; at baseline, patients had a median Mayo score of 9, with 86% of patients having moderate disease (Mayo score 6-10), and 14% having severe disease (Mayo score 11-12).
Concomitant immunomodulators or biologic therapies were not permitted.
The primary endpoint was clinical remission at Week 10, defined using a 3-component Mayo score without the physician global assessment: rectal bleeding subscore = 0, stool frequency subscore = 0 or 1 (and a decrease of ≥ 1 point from the baseline stool frequency subscore), and endoscopy subscore = 0 or 1 (an endoscopy subscore of 0 defined as normal or inactive disease, and an endoscopy subscore of 1 defined as presence of erythema, decreased vascular pattern and no friability).
The secondary endpoints were clinical response, endoscopic improvement, and endoscopic-histologic mucosal improvement. Clinical response (reduction from baseline in the 3-component Mayo score of ≥ 2 points and ≥ 35%, and a reduction from baseline in the rectal bleeding subscore of ≥ 1 point or an absolute rectal bleeding subscore of 0 or 1), endoscopic improvement (Mayo endoscopy subscore of 0 or 1), and endoscopic-histologic mucosal improvement [combined endoscopic improvement and histologic improvement of colonic tissue (no neutrophils in the epithelial crypts or lamina propria and no increase in eosinophils, no crypt destruction, and no erosions, ulcerations, or granulation tissue, i.e., Geboes < 2.0)].
A significantly greater proportion of patients treated with ZEPOSIA achieved clinical remission, clinical response, endoscopic improvement, and endoscopic-histologic mucosal improvement compared to placebo at Week 10 (see Table 9).
Table 9: Proportion of Patients Meeting Efficacy Endpoints in the Induction Period at Week 10 in UC Study 1 CI = confidence interval; TNF = tumor necrosis factor. a ZEPOSIA was initiated with a 7-day titration [see Dosage and Administration (2.2)]. b Treatment difference (adjusted for stratification factors of prior anti-TNF exposure and corticosteroid use at baseline). c Clinical remission is defined as: rectal bleeding subscore = 0, stool frequency subscore = 0 or 1 (and a decrease from baseline in the stool frequency subscore of ≥ 1 point), and endoscopy subscore = 0 or 1 without friability. d Clinical response is defined as a reduction from baseline in the 3-component Mayo score of ≥ 2 points and ≥ 35%, and a reduction from baseline in the rectal bleeding subscore of ≥ 1 point or an absolute rectal bleeding subscore of 0 or 1. e Endoscopic improvement is defined as a Mayo endoscopy subscore of 0 or 1 without friability. f Endoscopic-histologic mucosal improvement is defined as both Mayo endoscopic subscore of 0 or 1 without friability and histologic improvement of colonic tissue (defined as no neutrophils in the epithelial crypts or lamina propria and no increase in eosinophils, no crypt destruction, and no erosions, ulcerations, or granulation tissue, i.e., Geboes < 2.0). g p<0.0001. h p<0.001.
Endpoint
ZEPOSIA 0.92 mg Once Dailya (N=429)
Placebo (N=216)
Treatment Differenceb (95% CI)
n
%
n
%
Clinical remissionc
79
18%
13
6%
12% (8%, 17%)g
  Without prior TNF blocker exposure
66/299
22%
10/151
7%
  Prior TNF blocker exposure
13/130
10%
3/65
5%
Clinical responsed
205
48%
56
26%
22% (14%, 29%)g
  Without prior TNF blocker exposure
157/299
53%
44/151
29%
  Prior TNF blocker exposure
48/130
37%
12/65
19%
Endoscopic improvemente
117
27%
25
12%
16% (10%, 22%)g
  Without prior TNF blocker exposure
97/299
32%
18/151
12%
  Prior TNF blocker exposure
20/130
15%
7/65
11%
Endoscopic-histologic mucosal improvementf
54
13%
8
4%
9% (5%, 13%)h
  Without prior TNF blocker exposure
47/299
16%
6/151
4%
  Prior TNF blocker exposure
7/130
5%
2/65
3%
The relationship of endoscopic-histologic mucosal improvement, as defined in UC Study 1, at Week 10 to disease progression and long term outcomes was not evaluated during UC Study 1.
Rectal Bleeding Subscore and Stool Frequency Subscores
Decreases in rectal bleeding and stool frequency subscores were observed as early as Week 2 (i.e., 1 week after completing the required 7-day dosage titration) in patients treated with ZEPOSIA.
UC Study 2
In UC Study 2, a total of 457 patients who received ZEPOSIA in either UC Study 1 or in an open-label arm and achieved clinical response at Week 10 were re-randomized 1:1 and were treated with either ZEPOSIA 0.92 mg (n=230) or placebo (n=227) for 42 weeks (UC Study 2), for a total of 52 weeks of treatment.
Patients were permitted to be on stable doses of oral aminosalicylates. Corticosteroid tapering was required upon entering this study for patients who were receiving corticosteroids during the induction period. Concomitant oral immunomodulators or biologic therapies were not permitted. At study entry, 35% of patients were in clinical remission; 29% of patients were on corticosteroids; and 31% of patients had an inadequate response, loss of response, or intolerance to TNF blockers.
The primary endpoint was the proportion of patients in clinical remission at Week 52. The secondary endpoints at Week 52 were the proportion of patients with clinical response, endoscopic improvement, endoscopic-histologic mucosal improvement, corticosteroid-free clinical remission, and maintenance of clinical remission at Week 52 among patients who achieved clinical remission at Week 10 in UC Study 1.
The results of the efficacy endpoints in the maintenance period are shown in Table 10.
Table 10: Proportion of Patients Meeting Efficacy Endpoints in the Maintenance Period at Week 52 in UC Study 2 CI = confidence interval; TNF = tumor necrosis factor. a ZEPOSIA was initiated with a 7-day titration [see Dosage and Administration (2.2)]. b Treatment difference (adjusted for stratification factors of clinical remission and concomitant corticosteroid use at Week 10). c Clinical remission is defined as: rectal bleeding subscore = 0, stool frequency subscore = 0 or 1 (and a decrease from baseline in the stool frequency subscore of ≥ 1 point), and endoscopy subscore = 0 or 1 without friability. d Clinical response is defined as a reduction from baseline in the 3-component Mayo score of ≥ 2 points and ≥ 35%, and a reduction from baseline in the rectal bleeding subscore of ≥ 1 point or an absolute rectal bleeding subscore of 0 or 1. e Endoscopic improvement is defined as a Mayo endoscopy subscore of 0 or 1 without friability. f Maintenance of remission is defined as clinical remission at Week 52 in the subset of patients in clinical remission at Week 10. g Corticosteroid-free remission is defined as clinical remission at Week 52 while off corticosteroids for ≥ 12 weeks. h Endoscopic-histologic mucosal improvement is defined as both Mayo endoscopic score of 0 or 1 without friability and histologic improvement of colonic tissue (defined as no neutrophils in the epithelial crypts or lamina propria and no increase in eosinophils, no crypt destruction, and no erosions, ulcerations, or granulation tissue, i.e., Geboes <2.0). i p<0.0001. j p<0.001. k p=0.0025.
Endpoint
ZEPOSIA 0.92 mg Once Dailya (N=230)
Placebo (N=227)
Treatment Differenceb (95% CI)
Â
n
%
n
%
Clinical remissionc
85
37%
42
19%
19% (11%, 26%)i
  Without prior TNF blocker exposure
63/154
41%
35/158
22%
  Prior TNF blocker exposure
22/76
29%
7/69
10%
Clinical responsed
138
60%
93
41%
19% (10%, 28%)i
  Without prior TNF blocker exposure
96/154
62%
76/158
48%
  Prior TNF blocker exposure
42/76
55%
17/69
25%
Endoscopic improvemente
105
46%
60
26%
19% (11%, 28%)j
  Without prior TNF blocker exposure
77/154
50%
48/158
30%
  Prior TNF blocker exposure
28/76
37%
12/69
17%
Maintenance of clinical remission at Week 52 in the subset of patients in remission at Week 10f
41/79
52%
22/75
29%
24% (9%, 39%)k
  Without prior TNF blocker exposure
37/64
58%
19/58
33%
  Prior TNF blocker exposure
4/15
27%
3/17
18%
Corticosteroid-free clinical remissiong
73
32%
38
17%
15% (8%, 23%)j
  Without prior TNF blocker exposure
55/154
36%
31/158
20%
  Prior TNF blocker exposure
18/76
24%
7/69
10%
Endoscopic-histologic mucosal improvementh
68
30%
32
14%
16% (8%, 23%)j
  Without prior TNF blocker exposure
51/154
33%
28/158
18%
  Prior TNF blocker exposure
17/76
22%
4/69
6%
The relationship of endoscopic-histologic mucosal improvement, as defined in UC Study 2, at Week 52 to disease progression and long term outcomes was not evaluated during UC Study 2.
16 How Supplied/storage And Handling
16.1How Supplied
ZEPOSIA is available as capsules in the following dosage strengths:
• 0.23 mg ozanimod: light grey opaque body/light grey opaque cap imprinted with black ink "OZA" on the cap and "0.23 mg" on the body• 0.46 mg ozanimod: light grey opaque body/orange opaque cap imprinted with black ink "OZA" on the cap and "0.46 mg" on the body• 0.92 mg ozanimod: orange opaque body/orange opaque cap imprinted with black ink "OZA" on the cap and "0.92 mg" on the body
Capsules are supplied in the following strengths and package configurations:
Package configuration
Tablet strength
NDC number
Bottles of 30
0.92 mg ozanimod
59572-820-30
7-Day Starter Pack
7-capsule starter pack containing: (4) 0.23 mg ozanimod capsules and (3) 0.46 ozanimod mg capsules
59572-810-07
Starter Kit(7-Day Starter Pack and 0.92 mg 30-count Bottle)
37-capsule starter kit including:one 7-capsule starter pack containing:(4) 0.23 mg ozanimod capsules and(3) 0.46 mg ozanimod capsules andone bottle containing: (30) 0.92 mg ozanimod capsules
59572-890-91 59572-890-07 59572-890-30
Starter Kit
(7-Day Starter Pack and 0.92 mg 21-count Bottle)
28-capsule starter kit including:one 7-capsule starter pack containing:(4) 0.23 mg ozanimod capsules and(3) 0.46 mg ozanimod capsules andone bottle containing: (21) 0.92 mg ozanimod capsules
59572-890-28 59572-890-07 59572-890-21
16.2 Storage
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Risk of Infections
Inform patients that they may be more likely to get infections, some of which could be life-threatening, when taking ZEPOSIA and for 3 months after stopping it, and that they should contact their healthcare provider if they develop symptoms of infection [see Warnings and Precautions (5.1)]. Inform patients that prior or concomitant use of drugs that suppress the immune system may increase the risk of infection. Advise patients that some vaccines containing live virus (live attenuated vaccines) should be avoided during treatment with ZEPOSIA. Advise patients that if immunizations are planned, they should be administered at least 1 month prior to initiation of ZEPOSIA. Inform patients that the use of live attenuated vaccines should be avoided during and for 3 months after treatment with ZEPOSIA.
Progressive Multifocal Leukoencephalopathy
Inform patients that cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients who received ZEPOSIA and other S1P receptor modulators. Inform the patient that PML is characterized by a progression of deficits and usually leads to death or severe disability over weeks or months. Instruct the patient of the importance of contacting their healthcare provider if they develop any symptoms suggestive of PML. Inform the patient that typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes [see Warnings and Precautions (5.2)].
Cardiac Effects
Advise patients that initiation of ZEPOSIA treatment may result in a transient decrease in heart rate. Inform patients that to reduce this effect, dose titration is required. Advise patients that the dose titration is also required if a dose is missed for 1 day or more during the first 14 days of treatment [see Dosage and Administration (2.2, 2.4) and Warnings and Precautions (5.3)].
Liver Injury
Inform patients that ZEPOSIA may increase liver enzymes. Advise patients that they should contact their healthcare provider if they have any unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine [see Warnings and Precautions (5.4) ].
Pregnancy and Fetal Risk
Inform patients that, based on animal studies, ZEPOSIA may cause fetal harm. Discuss with women of childbearing age whether they are pregnant, might be pregnant, or are trying to become pregnant. Advise women of childbearing potential of the need for effective contraception during treatment with ZEPOSIA and for 3 months after stopping ZEPOSIA. Advise a female patient to immediately inform her healthcare provider if she is pregnant or planning to become pregnant [see Warnings and Precautions (5.5) and Use in Specific Populations (8.3)].
Pregnancy Registry
Encourage multiple sclerosis patients to enroll in the ZEPOSIA Pregnancy Registry if they become pregnant while taking ZEPOSIA [see Use in Specific Populations (8.1)].
Respiratory Effects
Advise patients that they should contact their healthcare provider if they experience new onset or worsening dyspnea [see Warnings and Precautions (5.7) ].
Macular Edema
Advise patients that ZEPOSIA may cause macular edema, and that they should contact their healthcare provider if they experience any changes in their vision. Inform patient with diabetes mellitus or a history of uveitis that their risk of macular edema may be increased [see Warnings and Precautions (5.8) ].
Posterior Reversible Encephalopathy Syndrome
Advise patients to immediately report to their healthcare provide any symptoms involving sudden onset of severe headache, altered mental status, visual disturbances, or seizure. Inform patients that delayed treatment could lead to permanent neurological consequences [see Warnings and Precautions (5.9) ].
Severe Increase in Multiple Sclerosis Disability after Stopping ZEPOSIA
Inform patients with multiple sclerosis that severe increase in disability has been reported after discontinuation of a S1P receptor modulator like ZEPOSIA. Advise patients to contact their physician if they develop worsening symptoms of MS following discontinuation of ZEPOSIA [see Warnings and Precautions (5.11) ].
Immune System Effects after Stopping ZEPOSIA
Advise patients that ZEPOSIA continues to have effects, such as lowering effects on peripheral lymphocyte count, for up to 3 months after the last dose [see Warnings and Precautions (5.12) ].
Marketed by:Bristol-Myers Squibb CompanyPrinceton, NJ 08543 USA
ZEPOSIA® is a trademark of Celgene Corporation, a Bristol-Myers Squibb company.
ZEPPI.010/ZEPMG.007
Spl Medguide Section
MEDICATION GUIDE ZEPOSIA® (zeh-poe'-see-ah)(ozanimod) capsules, for oral use
Read this Medication Guide before you start taking ZEPOSIA and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about ZEPOSIA? ZEPOSIA may cause serious side effects, including:
1. Infections. ZEPOSIA can increase your risk of serious infections that can be life-threatening and cause death. ZEPOSIA lowers the number of white blood cells (lymphocytes) in your blood. This will usually go back to normal within 3 months of stopping treatment. Your healthcare provider may do a blood test of your white blood cells before you start taking ZEPOSIA. Call your healthcare provider right away if you have any of the following symptoms of an infection during treatment with ZEPOSIA and for 3 months after your last dose of ZEPOSIA:
o fevero feeling very tiredo flu-like symptomso cougho painful and frequent urination (signs of a urinary tract infection)o rash
o headache with fever, neck stiffness, sensitivity to light, nausea or confusion (these may be symptoms of meningitis, an infection of the lining around your brain and spine)
Your healthcare provider may delay starting or may stop your ZEPOSIA treatment if you have an infection.
2. Progressive multifocal leukoencephalopathy (PML). ZEPOSIA can increase your risk for PML, which is a rare brain infection that usually leads to death or severe disability. If PML happens, it usually happens in people with weakened immune systems but has happened in people who do not have weakened immune systems. Symptoms of PML get worse over days to weeks. Call your doctor right away if you have any new or worsening symptoms of PML that have lasted several days, including:
o weakness on 1 side of your bodyo loss of coordination in your arms or legso decreased strengtho problems with balance
o changes in your visiono changes in your thinking or memoryo confusiono changes in your personality
3. Slow heart rate (also known as bradyarrhythmia) when you start taking ZEPOSIA. ZEPOSIA may cause your heart rate to temporarily slow down, especially during the first 8 days that you take ZEPOSIA. You will have a test to check the electrical activity of your heart called an electrocardiogram (ECG) before you take your first dose of ZEPOSIA. Call your healthcare provider if you experience the following symptoms of slow heart rate:
o dizzinesso lightheadednesso feeling like your heart is beating slowly or skipping beats
o shortness of breatho confusiono chest paino tiredness
Follow directions from your healthcare provider when starting ZEPOSIA and when you miss a dose. See "How should I take ZEPOSIA?".
See "What are possible side effects of ZEPOSIA?" for more information about side effects.
What is ZEPOSIA? ZEPOSIA is a prescription medicine used to treat:
• adults with relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease.• adults with moderately to severely active ulcerative colitis.
It is not known if ZEPOSIA is safe and effective in children.
Do not take ZEPOSIA if you:
• have had a heart attack, chest pain (unstable angina), stroke or mini-stroke (transient ischemic attack or TIA), or certain types of heart failure in the last 6 months.• have or have had a history of certain types of an irregular or abnormal heartbeat (arrhythmia) that is not corrected by a pacemaker.• have untreated, severe breathing problems during your sleep (sleep apnea).• take certain medicines called monoamine oxidase (MAO) inhibitors (such as selegiline, phenelzine, linezolid).
Talk to your healthcare provider before taking ZEPOSIA if you have any of these conditions or do not know if you have any of these conditions.
Before taking ZEPOSIA, tell your healthcare provider about all of your medical conditions, including if you:
• have a fever or infection, or you are unable to fight infections due to a disease, or take or have taken medicines that lower your immune system.• received a vaccine in the past 30 days or are scheduled to receive a vaccine. ZEPOSIA may cause vaccines to be less effective.• Before you start treatment with ZEPOSIA, your healthcare provider may give you a chicken pox (Varicella Zoster Virus) vaccine if you have not had one before.• have had chickenpox or have received the vaccine for chickenpox. Your healthcare provider may do a blood test for the chickenpox virus. You may need to get the full course of the vaccine for chickenpox and then wait 1 month before you start taking ZEPOSIA.• have a slow heart rate.• have an irregular or abnormal heartbeat (arrhythmia).• have a history of a stroke.• have heart problems, including a heart attack or chest pain.• have high blood pressure.• have liver problems.• have breathing problems, including during your sleep.• have eye problems, especially an inflammation of the eye called uveitis.• have diabetes.• are pregnant or plan to become pregnant. ZEPOSIA may harm your unborn baby. Talk with your healthcare provider if you are pregnant or plan to become pregnant. If you are a female who can become pregnant, you should use effective birth control during your treatment with ZEPOSIA and for 3 months after you stop taking ZEPOSIA. Talk with your healthcare provider about what birth control method is right for you during this time. Tell your healthcare provider right away if you become pregnant while taking ZEPOSIA or if you become pregnant within 3 months after you stop taking ZEPOSIA. Pregnancy Registry for MS patients: There is a pregnancy registry for women with multiple sclerosis who become pregnant during treatment with ZEPOSIA. If you become pregnant while taking ZEPOSIA, tell your healthcare provider right away. Talk to your healthcare provider about registering with the ZEPOSIA Pregnancy Registry. The purpose of this registry is to collect information about your health and your baby's health. Either you or your healthcare provider can enroll you in this registry by calling 1-877-301-9314 or visiting https://www.zeposiapregnancyregistry.com/.Information regarding registration of pregnant women with UC will be made available in the future.• are breastfeeding or plan to breastfeed. It is not known if ZEPOSIA passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take ZEPOSIA.
Tell your healthcare provider about all the medicines you take or have recently taken, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Using ZEPOSIA with other medicines can cause serious side effects. Especially tell your healthcare provider if you take or have taken:
• medicines that affect your immune system, such as alemtuzumab• medicines to control your heart rhythm (antiarrhythmics), or heart beat• CYP2C8 inducers such as rifampin• CYP2C8 inhibitors such as gemfibrozil (medicine to treat high fat in your blood)• opioids (pain medicine)• medicines to treat depression• medicines to treat Parkinson's disease• medicines to control your heart rate and blood pressure (beta blocker medicines and calcium channel blocker medicines)
You should not receive live vaccines during treatment with ZEPOSIA, for at least 1 month before taking ZEPOSIA and for 3 months after you stop taking ZEPOSIA. Vaccines may not work as well when given during treatment with ZEPOSIA.Talk with your healthcare provider if you are not sure if you take any of these medicines.Know the medicines you take. Keep a ul of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take ZEPOSIA? You will receive a 7-day starter pack. You must start ZEPOSIA by slowly increasing doses over the first week. Follow the dose schedule in the table below. This may reduce the risk of slowing of the heart rate.
Days 1-4
Take 0.23 mg (capsule in light grey color) 1 time a day
Days 5-7
Take 0.46 mg (capsule in half-light grey and half-orange color) 1 time a day
Days 8 and thereafter
Take 0.92 mg (capsule in orange color) 1 time a day, or as directed by your healthcare provider
• Take ZEPOSIA exactly as your healthcare provider tells you to take it.• Swallow ZEPOSIA capsules whole.• Take ZEPOSIA with or without food.• Avoid certain foods that are high (over 150 mg) in tyramine such as aged, fermented, cured, smoked and pickled foods. Eating these foods while taking ZEPOSIA may increase your blood pressure.• Do not stop taking ZEPOSIA without talking with your healthcare provider first.• Do not skip a dose.• Start taking ZEPOSIA with a 7-day starter pack.• If you miss 1 or more days of your ZEPOSIA dose during the first 14 days of treatment, talk to your healthcare provider. You will need to begin with another ZEPOSIA 7-day starter pack.• If you miss a dose of ZEPOSIA after the first 14 days of treatment, take the next scheduled dose the following day.
What are the possible side effects of ZEPOSIA? ZEPOSIA may cause serious side effects, including:
• See "What is the most important information I should know about ZEPOSIA?"• liver problems. ZEPOSIA may cause liver problems. Your healthcare provider will do blood tests to check your liver before you start taking ZEPOSIA. Call your healthcare provider right away if you have any of the following symptoms:
o unexplained nauseao vomitingo stomach area (abdominal) paino tiredness
o loss of appetiteo yellowing of the whites of your eyes or skino dark colored urine
• increased blood pressure. Your healthcare provider should check your blood pressure during treatment with ZEPOSIA. A sudden, severe increase in blood pressure (hypertensive crisis) can happen when you eat certain foods that contain high levels of tyramine. See "How should I take ZEPOSIA?" section for more information.• breathing problems. Some people who take ZEPOSIA have shortness of breath. Call your healthcare provider right away if you have new or worsening breathing problems.• a problem with your vision called macular edema. Your risk for macular edema is higher if you have diabetes or have had an inflammation of your eye called uveitis. Your healthcare provider should test your vision before you start taking ZEPOSIA if you are at higher risk for macular edema or at any time you notice vision changes during treatment with ZEPOSIA. Call your healthcare provider right away if you have any of the following symptoms:
o blurriness or shadows in the center of your visiono sensitivity to light
o a blind spot in the center of your visiono unusually colored vision
• swelling and narrowing of blood vessels in your brain. A condition called PRES (Posterior Reversible Encephalopathy Syndrome) is a rare condition that has happened with ZEPOSIA and with drugs in the same class. Symptoms of PRES usually get better when you stop taking ZEPOSIA. If left untreated, it may lead to a stroke. Your healthcare provider will do a test if you have any symptoms of PRES. Call your healthcare provider right away if you have any of the following symptoms:
o sudden severe headacheo sudden confusion
o sudden loss of vision or other changes in your visiono seizure
• severe worsening of multiple sclerosis (MS) after stopping ZEPOSIA. When ZEPOSIA is stopped, symptoms of MS may return and become worse compared to before or during treatment. Always talk to your healthcare provider before you stop taking ZEPOSIA for any reason. Tell your healthcare provider if you have worsening symptoms of MS after stopping ZEPOSIA.
The most common side effects of ZEPOSIA can include:
• upper respiratory tract infections• elevated liver enzymes
• low blood pressure when you stand up (orthostatic hypotension)• painful and frequent urination (signs of urinary tract infection)
• back pain• high blood pressure
• headache
These are not all of the possible side effects of ZEPOSIA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ZEPOSIA?
• Store ZEPOSIA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep ZEPOSIA and all medicines out of the reach of children.
General information about the safe and effective use of ZEPOSIA. Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not take ZEPOSIA for conditions for which it was not prescribed. Do not give ZEPOSIA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZEPOSIA that is written for health professionals.
What are the ingredients in ZEPOSIA? Active ingredient: ozanimod Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, and microcrystalline cellulose.The capsule shell contains: black iron oxide, gelatin, red iron oxide, titanium dioxide, and yellow iron oxide. Marketed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.ZEPOSIA® is a trademark of Celgene Corporation, a Bristol-Myers Squibb company.
This Medication Guide has been approved by the U.S. Food and Drug Administration.Revised: 8/2023
ZEPMG.007
Principal Display Panel - 0.92 Mg Capsule Bottle Label
NDC 59572-820-30
ZEPOSIA® (ozanimod) capsules
0.92 mg
Dispense the accompanyingMedication Guide to each patient.
Rx only30 Capsules
Bristol-Myers Squibb

Principal Display Panel - 7 Capsule Starter Pack Kit Carton
NDC 59572-810-07 Rx only
ZEPOSIA® (ozanimod) capsules
7-DAY STARTER PACK
This pack contains 7 capsules for dosing over 7 days. The contents of this pack are as follows:
Four 0.23 mg capsules Three 0.46 mg capsules
7 CAPSULES
Dispense the accompanying Medication Guide to each patient.
Bristol-Myers Squibb

Principal Display Panel - Kit Carton
Rx only
NDC 59572-890-91
ZEPOSIA® (ozanimod) capsules
STARTER KIT
STEP 1: (Days 1-7)
Starting at Day 1, complete the 7-Day Starter PackDays 1-4: Take one 0.23 mg capsule orally once dailyDays 5-7: Take one 0.46 mg capsule orally once daily
STEP 2: (Day 8 and thereafter)
After completing the 7-Day Starter Pack, begin takingone 0.92 mg capsule once daily or as directed from the 30-count bottle.
This Starter Kit contains 37 capsules for titration over 7 days up to prescribed maintenance dose of one 0.92 mg capsule taken once daily or as directed. The contents of the Starter Kit, which are not to be sold separately, are as follows:
Four 0.23 mg capsulesThree 0.46 mg capsulesThirty 0.92 mg capsules
37 CAPSULES
Dispense the accompanying Medication Guide to each patient.
Bristol Myers Squibb

Package/label Display Panel
Rx only
NDC 59572-890-28
ZEPOSIA® (ozanimod) capsules
STARTER KIT
STEP 1: (Days 1-7)
Starting at Day 1, complete the 7-Day Starter PackDays 1-4: Take one 0.23 mg capsule orally once dailyDays 5-7: Take one 0.46 mg capsule orally once daily
STEP 2: (Day 8 and thereafter)
After completing the 7-Day Starter Pack, begintaking one 0.92 mg capsule a day or as directedfrom the 21-count bottle.
This Starter Kit contains 28 capsules for titration over 7 days up to prescribed maintenance dose of one 0.92 mg capsule taken once daily or asdirected. The contents of the Starter Kit, whichare not to be sold separately, are as follows:
Four 0.23 mg capsulesThree 0.46 mg capsulesTwenty One 0.92 mg capsules
28 CAPSULES
Dispense the accompanying Medication Guide to each patient.
Bristol Myers Squibb

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
