UPTRAVI (selexipag 800 ug) Dailymed
Generic: selexipag is used for the treatment of Hypertension, Pulmonary

Go PRO for all pill images
Recent Major Changes Section
Contraindications ( 4 )10/2021
1 Indications And Usage
- UPTRAVI is a prostacyclin receptor agonist indicated for the treatment of pulmonary arterial hypertension (PAH, WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH. (
1.1 )1.1Pulmonary Arterial Hypertension
UPTRAVI is indicated for the treatment of pulmonary arterial hypertension (PAH, WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH.
Effectiveness of UPTRAVI tablets was established in a long-term study in PAH patients with WHO Functional Class II‚ÄďIII symptoms.
Patients had idiopathic and heritable PAH (58%), PAH associated with connective tissue disease (29%), PAH associated with congenital heart disease with repaired shunts (10%) [see Clinical Studies (14.1)] .
2 Dosage And Administration
- UPTRAVI tablets starting dose: 200 mcg twice daily. (
2.1 )- Increase the dose by 200 mcg twice daily at weekly intervals to the highest tolerated dose up to 1600 mcg twice daily. (
2.1 )- Maintenance dose is determined by tolerability. (
2.1 )- Moderate hepatic impairment: Starting dose 200 mcg once daily, increase the dose by 200 mcg once daily at weekly intervals to the highest tolerated dose up to 1600 mcg. (
2.5 )- UPTRAVI for injection dose is determined by the patient's current dose of UPTRAVI tablets. Administer UPTRAVI for injection by intravenous infusion, twice daily. (
2.2 )
See Full Prescribing Information for instructions on preparation and administration. (2.3 ).
2.1Recommended Dosage
UPTRAVI Film-coated Tablets
The recommended starting dosage of UPTRAVI tablets is 200 micrograms (mcg) given twice daily. Tolerability may be improved when taken with food [see Clinical Pharmacology (12.3)] .
Increase the dose in increments of 200 mcg twice daily, usually at weekly intervals, to the highest tolerated dose up to
1600 mcg twice daily. If a patient reaches a dose that cannot be tolerated, the dose should be reduced to the previous tolerated dose.
Do not split, crush, or chew tablets.
UPTRAVI for Injection
Use UPTRAVI for injection in patients who are temporarily unable to take oral therapy.
Administer UPTRAVI for injection twice daily by intravenous infusion at a dose that corresponds to the patient's current dose of UPTRAVI tablets (see Table 1). Administer UPTRAVI for injection as an 80-minute intravenous infusion.
2.2Preparation Instructions
Reconstitute and further dilute UPTRAVI for injection prior to intravenous infusion following aseptic procedures.
Determine the dose and total volume of reconstituted UPTRAVI solution required (see Table 1).
Reconstitution
- Remove the carton of UPTRAVI for injection from the refrigerator and allow to stand for approximately 30 to 60 minutes to reach room temperature (20¬įC to 25¬įC [68¬įF to 77¬įF]).
- The vial needs to be protected from light at all times. Ensure the protective wrap around label is covering the entire vial.
- Peel back light protective wrap on vial to inspect the contents in the vial. It should appear white to almost white broken cake or powdered material. Immediately close the light protective wrap on the vial.
 
- Reconstitute UPTRAVI for injection using a polypropylene syringe with 8.6 mL of 0.9% Sodium Chloride Injection, USP and slowly inject into the UPTRAVI vial with the stream directed toward the inside wall of the vial to obtain a concentration of 225 mcg/mL of selexipag.
- Document date and time of first puncture. Complete infusion within 4 hours of first puncture.
- Gently invert the vial and repeat until powder is completely dissolved. Do not shake.
- Inspect the vial by peeling back the light protective wrap around label for discoloration. The reconstituted solution should appear clear, colorless and free from foreign material. Do not use if the reconstituted solution is discolored, cloudy, or contains visible particles.
Dilution
- UPTRAVI for injection must be diluted in glass containers only.
- Withdraw 100 mL of 0.9% Sodium Chloride Injection, USP and transfer into an empty sterile glass container.
- Withdraw the required volume of reconstituted solution (see Table 1 for reconstituted transfer volume) from the UPTRAVI vial using a single, appropriately sized polypropylene syringe and dilute into the glass container containing 100 mL 0.9% Sodium Chloride Injection, USP to obtain the desired final dose.
- Mix the diluted UPTRAVI infusion solution by gentle inversion of the glass container 5 times. Do not shake.
- Protect diluted UPTRAVI infusion solution from light at all times. Assign a 4-hour expiry from the time of first vial puncture and wrap the glass container completely with light protective cover.
- The UPTRAVI infusion solution should be kept at room temperature (20¬įC‚Äď25¬įC [68¬įF‚Äď77¬įF]) and must be infused within 4 hours from the first puncture of the vial stopper (including infusion time).
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. The diluted UPTRAVI infusion solution should be clear and colorless. Discard if particulate matter is observed.
UPTRAVI for injection vials are single-dose, for single administration. All remaining reconstituted product must be discarded.
Table 1 Dosing Table for UPTRAVI intravenous based on current UPTRAVI tablets dose UPTRAVI tablets dose (mcg) for twice daily dosing Corresponding IV UPTRAVI Dose (mcg) for twice daily dosing Reconstituted transfer volume (mL) for dilution 200 225 1.0 400 450 2.0 600 675 3.0 800 900 4.0 1000 1125 5.0 1200 1350 6.0 1400 1575 7.0 1600 1800 8.0 2.3Administration Instructions
Administer by intravenous infusion over 80 minutes using an infusion set made of DEHP-free polyvinyl chloride (PVC), natural latex rubber-free microbore tubing protected from light.
Do not use a filter for administration.
Once the solution for infusion glass container is empty, continue the infusion at the same rate with 0.9% saline to empty the remaining solution for infusion in the IV line, to ensure that the entire solution for infusion has been administered.
2.4Interruptions and Discontinuations
If a dose of UPTRAVI is missed, patients should take a missed dose as soon as possible unless the next dose is within the next 6 hours.
If treatment is missed for 3 days or more, restart UPTRAVI at a lower dose and then retitrate.
2.5Dosage Adjustment in Patients with Hepatic Impairment
No dose adjustment of UPTRAVI is necessary for patients with mild hepatic impairment (Child-Pugh class A).
For patients with moderate hepatic impairment (Child-Pugh class B), the starting dose of UPTRAVI tablets is 200 mcg once daily. Increase in increments of 200 mcg once daily at weekly intervals, as tolerated [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)] .
Avoid use of UPTRAVI in patients with severe hepatic impairment (Child-Pugh class C).
2.6Dosage Adjustment with Co-administration of Moderate CYP2C8 Inhibitors
When co-administered with moderate CYP2C8 inhibitors (e.g., clopidogrel, deferasirox and teriflunomide), reduce the dosing of UPTRAVI to once daily [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)] .
3 Dosage Forms And Strengths
UPTRAVI is available in the following presentations:
Film-Coated Tablets
- 200 mcg selexipag [Light yellow tablet debossed with 2]
- 400 mcg selexipag [Red tablet debossed with 4]
- 600 mcg selexipag [Light violet tablet debossed with 6]
- 800 mcg selexipag [Green tablet debossed with 8]
- 1000 mcg selexipag [Orange tablet debossed with 10]
- 1200 mcg selexipag [Dark violet tablet debossed with 12]
- 1400 mcg selexipag [Dark yellow tablet debossed with 14]
- 1600 mcg selexipag [Brown tablet debossed with 16]
UPTRAVI for Injection
- 1800 mcg selexipag [Lyophilized powder white to almost white broken cake or powdered material, supplied in a 10 mL single-dose glass vial]
- Tablets: 200 mcg, 400 mcg, 600 mcg, 800 mcg, 1000 mcg, 1200 mcg, 1400 mcg, 1600 mcg. (
3 )- For Injection: 1800 mcg of selexipag as a lyophilized powder in a single-dose vial for reconstitution and dilution. (
3 )
4 Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Concomitant use of strong inhibitors of CYP2C8 (e.g., gemfibrozil) [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)] .
Concomitant use with strong CYP2C8 inhibitors. (4 ,7.1 ,12.3 )
Hypersensitivity to the active substance or to any of the excipients. (4 )
5 Warnings And Precautions
Pulmonary edema in patients with pulmonary veno-occlusive disease. If confirmed, discontinue treatment. (5.1 )
5.1Pulmonary Edema with Pulmonary Veno-Occlusive Disease
Should signs of pulmonary edema occur, consider the possibility of associated pulmonary veno-occlusive disease. If confirmed, discontinue UPTRAVI.
6 Adverse Reactions
Adverse reactions occurring more frequently (‚Č•5%) on UPTRAVI compared to placebo are headache, diarrhea, jaw pain, nausea, myalgia, vomiting, pain in extremity, and flushing. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Janssen at 1-800-526-7736 (1-800-JANSSEN) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
UPTRAVI Tablets
The safety of UPTRAVI tablets has been evaluated in a long-term, placebo-controlled study enrolling 1,156 patients with symptomatic PAH (GRIPHON study) [see Clinical Studies (14)] . The exposure to UPTRAVI in this trial was up to 4.2 years with median duration of exposure of 1.4 years.
Table 2 presents adverse reactions more frequent on UPTRAVI tablets than on placebo by ‚Č•3%.
Table 2: Adverse Reactions UPTRAVI Placebo Adverse Reaction N=575 N=577 Headache 65% 32% Diarrhea 42% 18% Jaw pain 26% 6% Nausea 33% 18% Myalgia 16% 6% Vomiting 18% 9% Pain in extremity 17% 8% Flushing 12% 5% Arthralgia 11% 8% Anemia 8% 5% Decreased appetite 6% 3% Rash 11% 8%
These adverse reactions are more frequent during the dose titration phase.
Hyperthyroidism was observed in 1% (n=8) of patients on UPTRAVI tablets and in none of the patients on placebo.
UPTRAVI for Injection
Infusion-site reactions (infusion site erythema/redness, pain and swelling) were reported with UPTRAVI for Injection.
Laboratory Test Abnormalities
Hemoglobin
In a Phase 3 placebo-controlled study in patients with PAH, mean absolute changes in hemoglobin at regular visits compared to baseline ranged from ‚ąí0.34 to ‚ąí0.02 g/dL in the UPTRAVI group compared to ‚ąí0.05 to 0.25 g/dL in the placebo group. A decrease in hemoglobin concentration to below 10 g/dL was reported in 8.6% of patients treated with UPTRAVI tablets and 5.0% of placebo-treated patients.
Thyroid Function Tests
In a Phase 3 placebo-controlled study in patients with PAH, a reduction (up to ‚ąí0.3 MU/L from a baseline median of 2.5 MU/L) in median thyroid-stimulating hormone (TSH) was observed at most visits in the UPTRAVI group. In the placebo group, little change in median values was apparent. There were no mean changes in triiodothyronine or thyroxine in either group.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of UPTRAVI.
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular disorders: symptomatic hypotension
7 Drug Interactions
- Moderate CYP2C8 inhibitors (e.g., clopidogrel, deferasirox and teriflunomide) increase exposure to the active metabolite of UPTRAVI. Reduce the dosing of UPTRAVI to once daily (
2.6 ,7.1 ,12.3 ).- CYP2C8 inducers (e.g., rifampin) decrease exposure to the active metabolite. Increase up to twice the dose of UPTRAVI (
7.2 ,12.3 )7.1CYP2C8 Inhibitors
Concomitant administration with gemfibrozil, a strong inhibitor of CYP2C8, doubled the exposure to selexipag and increased exposure to the active metabolite by approximately 11-fold. Concomitant administration of UPTRAVI with strong inhibitors of CYP2C8 (e.g., gemfibrozil) is contraindicated [see Contraindications (4) and Clinical Pharmacology (12.3)] .
Concomitant administration of UPTRAVI tablets with clopidogrel, a moderate inhibitor of CYP2C8, had no relevant effect on the exposure to selexipag and increased the exposure to the active metabolite by approximately 2.7-fold [see Clinical Pharmacology (12.3)] . Reduce the dosing of UPTRAVI to once daily in patients on a moderate CYP2C8 inhibitor [see Dosage and Administration (2.6)] .
7.2CYP2C8 Inducers
Concomitant administration with an inducer of CYP2C8 and UGT 1A3 and 2B7 enzymes (rifampin) halved exposure to the active metabolite. Increase dose up to twice of UPTRAVI when co-administered with rifampin. Reduce UPTRAVI when rifampin is stopped [see Clinical Pharmacology (12.3)] .
8 Use In Specific Populations
- Nursing mothers: Discontinue UPTRAVI or breastfeeding. (
8.2 )- Severe hepatic impairment: Avoid use. (
8.6 )8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies with UPTRAVI in pregnant women. Animal reproduction studies performed with selexipag showed no clinically relevant effects on embryofetal development and survival. A slight reduction in maternal as well as in fetal body weight was observed when pregnant rats were administered selexipag during organogenesis at a dose producing an exposure to the active metabolite approximately 47 times that in humans at the maximum recommended human dose. No adverse developmental outcomes were observed with oral administration of selexipag to pregnant rabbits during organogenesis at exposures to the active metabolite up to 50 times the human exposure at the maximum recommended human dose.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2‚Äď4% and 15‚Äď20%, respectively.
Data
Animal Data
Pregnant rats were treated with selexipag using oral doses of 2, 6, and 20 mg/kg/day (up to 47 times the exposure to the active metabolite at the maximum recommended human oral dose of 1600 mcg twice daily on an area under the curve [AUC] basis) during the period of organogenesis (gestation days 7 to 17). Selexipag did not cause adverse developmental effects to the fetus in this study. A slight reduction in fetal body weight was observed in parallel with a slight reduction in maternal body weight at the high dose.
Pregnant rabbits were treated with selexipag using oral doses of 3, 10, and 30 mg/kg (up to 50 times the exposure to the active metabolite at the maximum recommended human oral dose of 1600 mcg twice daily on an AUC basis) during the period of organogenesis (gestation days 6 to 18). Selexipag did not cause adverse developmental effects to the fetus in this study.
In a pre- and post-natal development study, pregnant rats were treated with selexipag from gestation day 7 through lactation day 20 at oral doses of 2, 6, and 20 mg/kg/day (up to 35 times the exposure to the active metabolite at the maximum recommended human dose of 1600 mcg twice daily on an AUC basis). Treatment with selexipag did not cause adverse developmental effects in this study at any dose.
8.2 Lactation
It is not known if UPTRAVI is present in human milk. Selexipag or its metabolites were present in the milk of rats. Because many drugs are present in the human milk and because of the potential for serious adverse reactions in nursing infants, discontinue nursing or discontinue UPTRAVI.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1,368 subjects in clinical studies of UPTRAVI tablets, 248 subjects were 65 years of age and older, while 19 were 75 and older. No overall differences were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity cannot be ruled out.
8.6Patients with Hepatic Impairment
No adjustment to the dosing regimen is needed in patients with mild hepatic impairment (Child-Pugh class A).
A once-daily regimen is recommended in patients with moderate hepatic impairment (Child-Pugh class B) due to the increased exposure to selexipag and its active metabolite. There is no experience with UPTRAVI in patients with severe hepatic impairment (Child-Pugh class C). Avoid use of UPTRAVI in patients with severe hepatic impairment [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)] .
8.7Patients with Renal Impairment
No adjustment to the dosing regimen is needed in patients with estimated glomerular filtration rate >15 mL/min/1.73 m 2.
There is no clinical experience with UPTRAVI in patients undergoing dialysis or in patients with glomerular filtration rates <15 mL/min/1.73 m 2 [see Clinical Pharmacology (12.3)] .
10 Overdosage
Isolated cases of overdose with UPTRAVI tablets up to 3200 mcg were reported. Mild, transient nausea was the only reported consequence. In the event of overdose, supportive measures must be taken as required. Dialysis is unlikely to be effective because selexipag and its active metabolite are highly protein-bound.
11 Description
UPTRAVI contains selexipag, a prostacyclin receptor agonist. The chemical name of selexipag is 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}- N-(methylsulfonyl) acetamide. It has a molecular formula of C 26H 32N 4O 4S and a molecular weight of 496.62. Selexipag has the following structural formula:
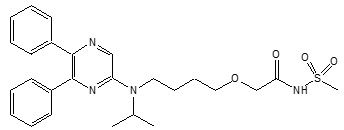
Selexipag is a pale yellow crystalline powder that is practically insoluble in water. In the solid state selexipag is very stable, is not hygroscopic, and is not light sensitive.
UPTRAVI ¬ģ (selexipag) tablets: depending on the dose strength, each round film-coated tablet for oral administration contains 200, 400, 600, 800, 1000, 1200, 1400, or 1600 mcg of selexipag. The tablets include the following inactive ingredients: corn starch, D-mannitol, hydroxypropyl cellulose, low substituted hydroxypropyl cellulose, and magnesium stearate. The tablets are film coated with a coating material containing carnauba wax, hypromellose, propylene glycol, titanium dioxide, along with mixtures of iron oxide black, iron oxide red or iron oxide yellow.
UPTRAVI ¬ģ (selexipag) for injection: contains 1800 mcg of selexipag per vial. UPTRAVI for injection includes the following inactive ingredients: glycine (180 mg), phosphoric acid (3.53 mg), polysorbate 20 (10.8 mg) and sodium hydroxide (for pH adjustment). UPTRAVI for injection is provided in 10 mL Type I clear glass vials closed by a stopper and tear-off aluminum seal.
12 Clinical Pharmacology
12.1 Mechanism of Action
Selexipag is a prostacyclin receptor (IP receptor) agonist that is structurally distinct from prostacyclin. Selexipag is hydrolyzed by carboxylesterase 1 to yield its active metabolite, which is approximately 37-fold as potent as selexipag. Selexipag and the active metabolite are selective for the IP receptor versus other prostanoid receptors (EP 1‚Äď4, DP, FP, and TP).
12.2 Pharmacodynamics
Cardiac Electrophysiology
At the maximum tolerated dose of 1600 mcg UPTRAVI tablets twice daily, UPTRAVI does not prolong the QT interval to any clinically relevant extent.
Platelet Aggregation
Both selexipag and its active metabolite caused concentration-dependent inhibition of platelet aggregation in vitro with an IC 50 of 5.5 ¬ĶM and 0.21 ¬ĶM, respectively. However, at clinically relevant concentrations, there was no effect on platelet aggregation test parameters as seen following multiple-dose administrations of UPTRAVI tablets in healthy subjects from 400 to 1800 mcg twice daily.
Pulmonary Hemodynamics
A Phase 2 clinical study assessed hemodynamic variables after 17 weeks of oral treatment in patients with PAH WHO Functional Class II‚ÄďIII and concomitantly receiving endothelin receptor antagonists (ERAs) and/or phosphodiesterase type 5 (PDE-5) inhibitors. Patients titrating UPTRAVI tablets to an individually tolerated dose (200 mcg twice daily increments up to 800 mcg twice daily) (N=33) achieved a statistically-significant mean reduction in pulmonary vascular resistance of 30.3% (95% confidence interval [CI] ‚ąí44.7%, ‚ąí12.2%) and an increase in cardiac index (median treatment effect) of 0.41 L/min/m 2 (95% CI 0.10, 0.71) compared to placebo (N=10).
Drug Interaction
In a study in healthy subjects, UPTRAVI tablets (400 mcg twice a day) did not influence the pharmacodynamic effect of warfarin on the international normalized ratio.
12.3 Pharmacokinetics
The pharmacokinetics of selexipag and its active metabolite have been studied primarily in healthy subjects. The pharmacokinetics of selexipag and the active metabolite, after both single- and multiple-dose oral administration, were dose-proportional up to a single dose of 800 mcg and multiple doses of up to 1800 mcg twice daily. The pharmacokinetics of selexipag and the active metabolite, after multiple-dose intravenous administration, were dose-proportional in the tested dose range from 450 to 1800 mcg twice a day.
In healthy subjects, inter-subject variability in exposure (area under the curve over a dosing interval, AUC) at steady-state following oral administration was 43% and 39% for selexipag and the active metabolite, respectively. Intra-subject variability in exposure was 24% and 19% for selexipag and the active metabolite, respectively.
Exposures to selexipag and the active metabolite at steady-state in PAH patients and healthy subjects were similar. The pharmacokinetics of selexipag and the active metabolite in PAH patients were not influenced by the severity of the disease and did not change with time.
The corresponding UPTRAVI tablets and UPTRAVI for injection doses (Table 1) provide similar exposure to the active metabolite in PAH patients at steady-state, whereas the exposure to selexipag is approximately twice as high after intravenous administration compared to oral administration.
Both in healthy subjects and PAH patients, after oral administration, exposure at steady-state to the active metabolite is approximately 3- to 4-fold that of selexipag.
Absorption
The absolute bioavailability of orally administered selexipag is approximately 49%. Upon oral administration, maximum observed plasma concentrations of selexipag and its active metabolite are reached within about 1‚Äď3 hours and 3‚Äď4 hours, respectively.
In the presence of food, the absorption of selexipag was prolonged resulting in a delayed time to peak concentration (T max) and ~30% lower peak plasma concentration (C max). The exposure to selexipag and the active metabolite (AUC) did not significantly change in the presence of food.
Distribution
The volume of distribution of selexipag at steady-state is 11.7 L.
Selexipag and its active metabolite are highly bound to plasma proteins (approximately 99% in total and to the same extent to albumin and alpha1-acid glycoprotein).
Metabolism
Selexipag is hydrolyzed to its active metabolite, (free carboxylic acid) in the liver and intestine by carboxylesterases. Oxidative metabolism, catalyzed mainly by CYP2C8 and to a smaller extent by CYP3A4, leads to the formation of hydroxylated and dealkylated products. UGT1A3 and UGT2B7 are involved in the glucuronidation of the active metabolite. Except for the active metabolite, none of the circulating metabolites in human plasma exceeds 3% of the total drug-related material.
Elimination
Elimination of selexipag is predominately via metabolism with a mean terminal half-life of 0.8‚Äď2.5 hours. The terminal half-life of the active metabolite is 6.2‚Äď13.5 hours. Selexipag does not accumulate following twice daily repeat administration. There is minimal accumulation of the active metabolite upon twice daily repeat administration suggesting that the effective half-life is in the range of 3‚Äď4 hours. The total body clearance of selexipag is 17.9 L/hour.
Excretion
In a study in healthy subjects with radiolabeled selexipag, approximately 93% of radioactive drug material was eliminated in feces and only 12% in urine. Neither selexipag nor its active metabolite were found in urine.
Specific Populations
No clinically relevant effects of sex, race, age or body weight on the pharmacokinetics of selexipag and its active metabolite have been observed in healthy subjects or PAH patients.
Age
The pharmacokinetic variables (C max and AUC) were similar in adult and elderly subjects up to 75 years of age. There was no effect of age on the pharmacokinetics of selexipag and the active metabolite in PAH patients.
Hepatic Impairment
In subjects with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment, exposure to selexipag was 2- and 4-fold that seen in healthy subjects. Exposure to the active metabolite of selexipag remained almost unchanged in subjects with mild hepatic impairment and was doubled in subjects with moderate hepatic impairment [see Use in Specific Populations (8.6)] .
Based on pharmacokinetic modeling of data from a study in subjects with hepatic impairment, the exposure to the active metabolite at steady-state in subjects with moderate hepatic impairment (Child-Pugh class B) after a once daily regimen is expected to be similar to that in healthy subjects receiving a twice daily regimen. The exposure to selexipag at steady-state in these patients during a once daily regimen is predicted to be approximately 2-fold that seen in healthy subjects receiving a twice-daily regimen.
Renal Impairment
A 40‚Äď70% increase in exposure (maximum plasma concentration and area under the plasma concentration-time curve) to selexipag and its active metabolite was observed in subjects with severe renal impairment (estimated glomerular filtration rate ‚Č•15 mL/min/1.73 m 2 and <30 mL/min/1.73 m 2) [see Use in Specific Populations (8.7)] .
Drug Interaction Studies
Drug interaction studies have been performed in adult subjects using UPTRAVI tablets.
In Vitro Studies
Selexipag is hydrolyzed to its active metabolite by carboxylesterases. Selexipag and its active metabolite both undergo oxidative metabolism mainly by CYP2C8 and to a smaller extent by CYP3A4. The glucuronidation of the active metabolite is catalyzed by UGT1A3 and UGT2B7. Selexipag and its active metabolite are substrates of OATP1B1 and OATP1B3. Selexipag is a substrate of P-gp, and the active metabolite is a substrate of the transporter of breast cancer resistance protein (BCRP).
Selexipag and its active metabolite do not inhibit or induce cytochrome P450 enzymes and transport proteins at clinically relevant concentrations.
The results of in vivo drug interaction studies are presented in Figure 1 and 2.
Figure 1 Effect of Other Drugs on Selexipag and its Active Metabolite
* ERA and PDE-5 inhibitor data from GRIPHON.
Figure 2 Effect of UPTRAVI on Other Drugs
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: In the 2-year carcinogenicity studies, chronic oral administration of selexipag revealed no evidence of carcinogenic potential in rats at 100 mg/kg/day and mice at 500 mg/kg/day which resulted in the exposures to the active metabolite more than 25 times the human exposure at the maximum recommended human oral dose of 1600 mcg twice daily on an AUC basis.
Mutagenesis: Selexipag and the active metabolite are not genotoxic on the basis of the overall evidence of conducted genotoxicity studies.
Fertility: In rats administered with selexipag orally, the no effect dose for effects on fertility was 60 mg/kg/day which resulted in the exposure to the active metabolite approximately 175 times the human exposure at the maximum recommended human oral dose of 1600 mcg twice daily on an AUC basis.
14 Clinical Studies
14.1Efficacy of UPTRAVI Tablets in Patients with Pulmonary Arterial Hypertension
The effect of UPTRAVI tablets on progression of PAH was demonstrated in a multi-center, double-blind, placebo-controlled, parallel group, event-driven study (GRIPHON) in 1,156 patients with symptomatic (WHO Functional Class I [0.8%], II [46%], III [53%], and IV [1%]) PAH. Patients were randomized to either placebo (N=582), or UPTRAVI tablets (N=574). The dose was increased in weekly intervals by increments of 200 mcg twice a day to the highest tolerated dose up to 1600 mcg twice a day.
The primary study endpoint was the time to first occurrence up to end-of-treatment of: a) death, b) hospitalization for PAH, c) PAH worsening resulting in need for lung transplantation, or balloon atrial septostomy, d) initiation of parenteral prostanoid therapy or chronic oxygen therapy, or e) other disease progression based on a 15% decrease from baseline in 6MWD plus worsening of Functional Class or need for additional PAH-specific therapy.
The mean age was 48 years, the majority of patients were white (65%) and female (80%). Nearly all patients were in WHO Functional Class II and III at baseline.
Idiopathic or heritable PAH was the most common etiology in the study population (58%) followed by PAH associated with connective tissue disease (29%), PAH associated with congenital heart disease with repaired shunts (10%), drugs and toxins (2%), and HIV (1%).
At baseline, the majority of enrolled patients (80%) were being treated with a stable dose of an endothelin receptor antagonist (15%), a PDE-5 inhibitor (32%), or both (33%).
Patients on UPTRAVI tablets achieved doses within the following groups: 200‚Äď400 mcg (23%), 600‚Äď1000 mcg (31%) and 1200‚Äď1600 mcg (43%).
Treatment with UPTRAVI tablets resulted in a 40% reduction (99% CI: 22 to 54%; two-sided log-rank p-value <0.0001) of the occurrence of primary endpoint events compared to placebo (Table 3; Figure 3). The beneficial effect of UPTRAVI was primarily attributable to a reduction in hospitalization for PAH and a reduction in other disease progression events (Table 3). The observed benefit of UPTRAVI was similar regardless of the dose achieved when patients were titrated to their highest tolerated dose [see Dosage and Administration (2.1)] .
Figure 3 Kaplan-Meier Estimates of the First Morbidity-Mortality Event in GRIPHON
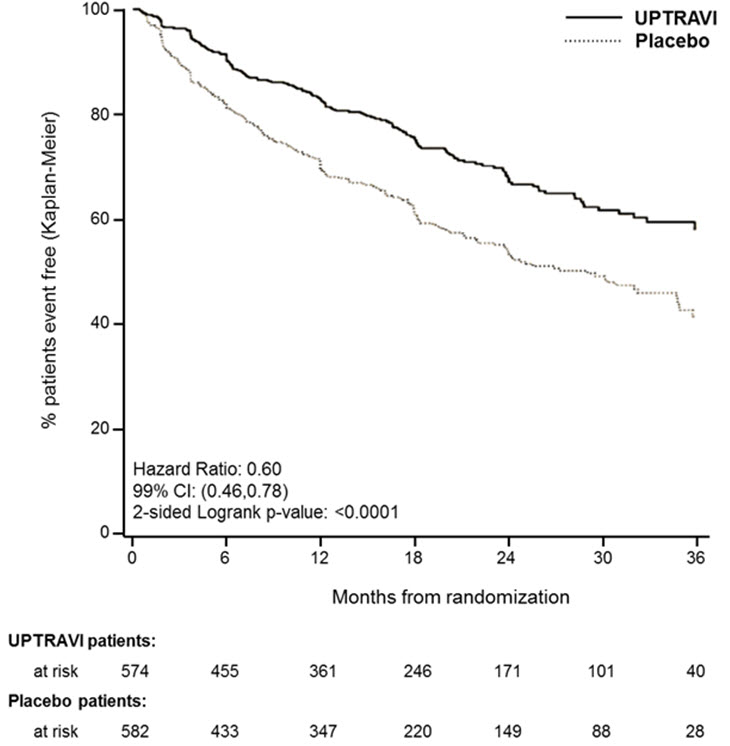
Table 3: Primary Endpoints and Related Components in GRIPHON UPTRAVI N=574 Placebo N=582 Hazard Ratio (99% CI) p-value n % n % Primary endpoint events up to the end of treatment All primary endpoint events As first event: 155 27.0 242 41.6 0.60 [0.46, 0.78] <0.0001
- Hospitalization for PAH
78 13.6 109 18.7
- Other disease progression (Decrease in 6MWD plus worsening functional class or need for other therapy)
38 6.6 100 17.2
- Death
28 4.9 18 3.1
- Parenteral prostanoid or chronic oxygen therapy
10 1.7 13 2.2
- PAH worsening resulting in need for lung transplantation or balloon atrial septostomy
1 0.2 2 0.3
It is not known if the excess number of deaths in the UPTRAVI group is drug-related because there were so few deaths and the imbalance was not observed until 18 months into GRIPHON.
Figures 4A, B, and C show time to first event analyses for primary endpoint components of hospitalization for PAH (A), other disease progression (B), and death (C) all censored 7 days after any primary end point event (because many patients on placebo transitioned to open-label UPTRAVI at this point).
Figure 4A Hospitalization for PAH as the First Endpoint in GRIPHON
Figure 4B Disease Progression as the First Endpoint in GRIPHON
Figure 4C Death as the First Endpoint in GRIPHON
The treatment effect of UPTRAVI on time to first primary event was consistent irrespective of background PAH therapy (i.e., in combination with an ERA, PDE-5i, both, or without background therapy) (Figure 5).
Figure 5 Subgroup Analyses of the Primary Endpoint in GRIPHON
Note: Race group "Other" is not displayed in analysis, as the population is less than 30. EU = Number of UPTRAVI patients with events, NU = Number of patients randomized to UPTRAVI, EP = Number of Placebo patients with events, NP = Number of patients randomized to Placebo, HR = Hazard Ratio, CI = Confidence Interval, the size of the squares represent the number of patients in the subgroup.
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all were pre-specified. The 99% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
6-Minute Walk Distance (6MWD)
Exercise capacity was evaluated as a secondary endpoint. Median absolute change from baseline to week 26 in 6MWD measured at trough (i.e., at approximately 12 hours post-dose) was +4 meters with UPTRAVI and -9 meters in the placebo group. This resulted in a placebo-corrected median treatment effect of 12 meters (99% CI: 1, 24 meters; two-sided p = 0.005).
Long-Term Treatment of PAH
In long-term follow-up of patients who were treated with UPTRAVI in the pivotal study and the open-label extension (N=574), Kaplan-Meier estimates of survival of these patients across the GRIPHON study and the long-term extension study at 1, 2, 5 and 7 years were 92%, 85%, 71%, and 63%, respectively. The median exposure to UPTRAVI was 3 years. These uncontrolled observations do not allow comparison with a control group not given UPTRAVI and cannot be used to determine the long-term effect of UPTRAVI on mortality.
16 How Supplied/storage And Handling
UPTRAVI ¬ģ (selexipag) film-coated, round tablets are supplied in the following configurations:
Strength (mcg) Color Debossing NDC-XXX Bottle of 60 NDC-XXX Bottle of 140 200 Light yellow 2 66215-602-06 66215-602-14 400 Red 4 66215-604-06 Not Available 600 Light violet 6 66215-606-06 Not Available 800 Green 8 66215-608-06 Not Available 1000 Orange 10 66215-610-06 Not Available 1200 Dark violet 12 66215-612-06 Not Available 1400 Dark yellow 14 66215-614-06 Not Available 1600 Brown 16 66215-616-06 Not Available
UPTRAVI ¬ģ (selexipag) tablets are also supplied in a Titration Pack [NDC 66215-628-20] that includes a 140-count bottle of 200-mcg tablets and a 60-count bottle of 800-mcg tablets.
STORAGE AND HANDLING SECTION
Store at 20¬įC to 25¬įC (68¬įF to 77¬įF). Excursions are permitted between 15¬įC and 30¬įC (59¬įF and 86¬įF) [see USP Controlled Room Temperature].
Keep out of reach of children.
UPTRAVI ¬ģ (selexipag) for injection, for intravenous use, is supplied in a 10 mL Type I glass vial closed by a stopper and sealed with an aluminum flip-off button, containing 1800 mcg of selexipag [NDC 66215-718-01].
UPTRAVI (selexipag) for injection is available in cartons containing 1 single-dose vial.
Storage conditions for UPTRAVI for injection: Store the original carton containing glass vial in a refrigerator at 2¬įC to 8¬įC (36¬ļF to 46¬ļF) until use in order to protect from light.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform Patients:
- To take a missed dose as soon as possible, unless the next dose is within the next 6 hours.
- Not to split, crush, or chew tablets.
Manufactured for:
Actelion Pharmaceuticals US, Inc. a Janssen Pharmaceutical Company Titusville, NJ 08560, USA
JN20220728
For patent information: www.janssenpatents.com
¬© 2015 ‚Äď 2021 Actelion Pharmaceuticals US, Inc.
Spl Patient Package Insert Section
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 07/2022 PATIENT INFORMATION UPTRAVI ¬ģ (up-TRA-vee) (selexipag) tablets UPTRAVI ¬ģ (up-TRA-vee) (selexipag) for injection What is UPTRAVI? It is not known if UPTRAVI is safe and effective in children.
- UPTRAVI is a prescription medicine used to treat pulmonary arterial hypertension (PAH) which is high blood pressure in the arteries of your lungs.
- UPTRAVI can help slow down the progression of your disease and lower your risk of being hospitalized for PAH.
Do not take UPTRAVI if you:
- take gemfibrozil because this medicine may affect how UPTRAVI works and cause side effects.
- are allergic to selexipag or any of the other ingredients of this medicine (uled under Inactive ingredients).
Before you take UPTRAVI, tell your healthcare provider about all of your medical conditions, including if you: Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. UPTRAVI and other medicines may affect each other causing side effects. Do not start any new medicine until you check with your healthcare provider.
- have liver problems.
- have narrowing of the pulmonary veins, a condition called pulmonary veno-occlusive disease.
- are pregnant or plan to become pregnant. It is not known if UPTRAVI will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if UPTRAVI passes into your breast milk. You and your healthcare provider should decide if you will take UPTRAVI or breastfeed. You should not do both.
How should I take UPTRAVI? UPTRAVI tablets UPTRAVI given by intravenous (IV) injection
- Take UPTRAVI exactly as your healthcare provider tells you to take it. Do not stop taking UPTRAVI unless your healthcare provider tells you to stop.
- Your healthcare provider will slowly increase your dose to find the dose of UPTRAVI that is right for you.
- If you have side effects, your healthcare provider may tell you to change your dose of UPTRAVI.
- UPTRAVI can be taken with or without food. Taking UPTRAVI with food may help you tolerate UPTRAVI better.
- UPTRAVI is usually taken 2 times each day.
- Swallow UPTRAVI tablets whole. Do not split, crush or chew UPTRAVI tablets.
- If you miss a dose of UPTRAVI, take it as soon as you remember. If your next scheduled dose is due within 6 hours, skip the missed dose. Take the next dose at your regular time.
- If you miss 3 or more days of UPTRAVI, call your healthcare provider to see if your dose needs to be changed.
- If you take too much UPTRAVI, call your healthcare provider or go to the nearest hospital emergency room right away.
- Your healthcare provider will give you UPTRAVI into your vein through an intravenous (IV) line.
- Your healthcare provider will decide how much UPTRAVI for injection you will receive each day, based on your current dose of UPTRAVI tablets.
What are the possible side effects of UPTRAVI? The most common side effects of UPTRAVI include:
- headache
- jaw pain
- muscle pain
- pain in arms or legs
- pain in joints
- decreased appetite
- diarrhea
- nausea
- vomiting
- flushing
- low red blood cell count
- rash
- pain, redness or swelling at the injection site for UPTRAVI for injection
These are not all of the possible side effects of UPTRAVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store UPTRAVI tablets? Store UPTRAVI tablets at room temperature between 68¬įF and 77¬įF (20¬įC and 25¬įC). Keep UPTRAVI and all medicines out of the reach of children. General information about the safe and effective use of UPTRAVI Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use UPTRAVI for a condition for which it was not prescribed. Do not give UPTRAVI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about UPTRAVI that is written for health professionals. What are the ingredients in UPTRAVI? UPTRAVI tablets Active ingredient: selexipag Inactive ingredients: corn starch, D-mannitol, hydroxypropyl cellulose, low substituted hydroxypropyl cellulose, and magnesium stearate. The tablets are film coated with a coating material containing carnauba wax, hypromellose, propylene glycol, titanium dioxide, along with mixtures of iron oxide black, iron oxide red, or iron oxide yellow. UPTRAVI for injection Active ingredient: selexipag Inactive ingredients: glycine, phosphoric acid, polysorbate 20, and sodium hydroxide. Manufactured for: Actelion Pharmaceuticals US, Inc. a Janssen Pharmaceutical Company Titusville, NJ 08560, USA JN20220728 For patent information: www.janssenpatents.com ¬© 2015 ‚Äď 2021 Actelion Pharmaceuticals US, Inc. For more information, contact Janssen at 1-800-526-7736 (1-800-JANSSEN) or go to www.UPTRAVI.com.
Principal Display Panel - 200 Mcg Tablet Bottle Carton
NDC 66215-602-06
Uptravi ¬ģ (selexipag) tablets
200 mcg
Rx only
60 film-coated tablets
janssen
Principal Display Panel - 400 Mcg Tablet Bottle Carton
NDC 66215-604-06
Uptravi ¬ģ (selexipag) tablets
400 mcg
Rx only
60 film-coated tablets
janssen
Principal Display Panel - 600 Mcg Tablet Bottle Carton
NDC 66215-606-06
Uptravi ¬ģ (selexipag) tablets
600 mcg
Rx only
60 film-coated tablets
janssen
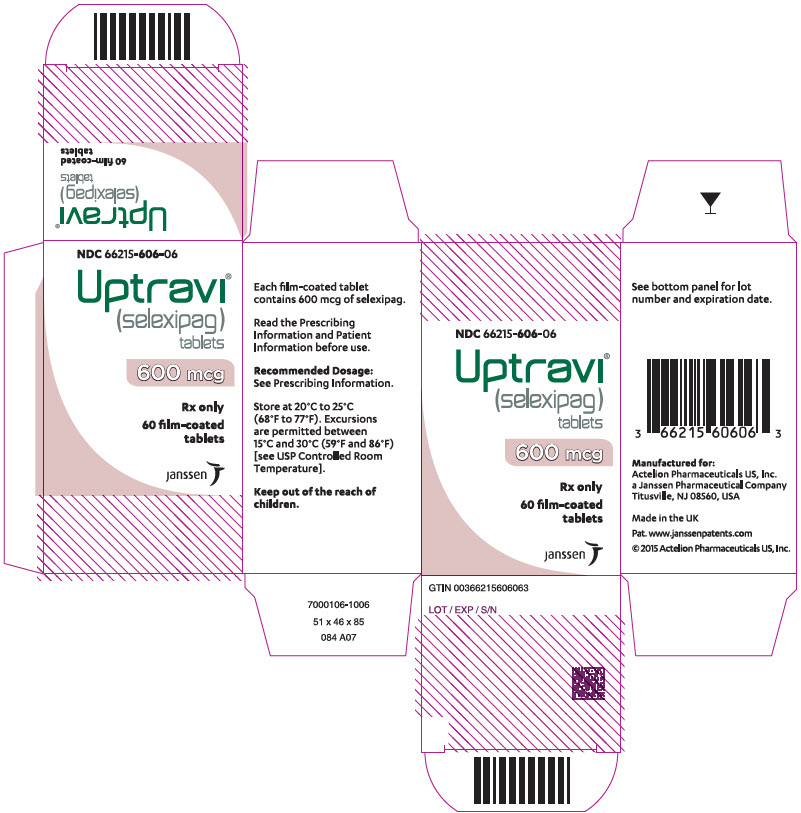
Principal Display Panel - 800 Mcg Tablet Bottle Carton
NDC 66215-608-06
Uptravi ¬ģ (selexipag) tablets
800 mcg
Rx only
60 film-coated tablets
janssen
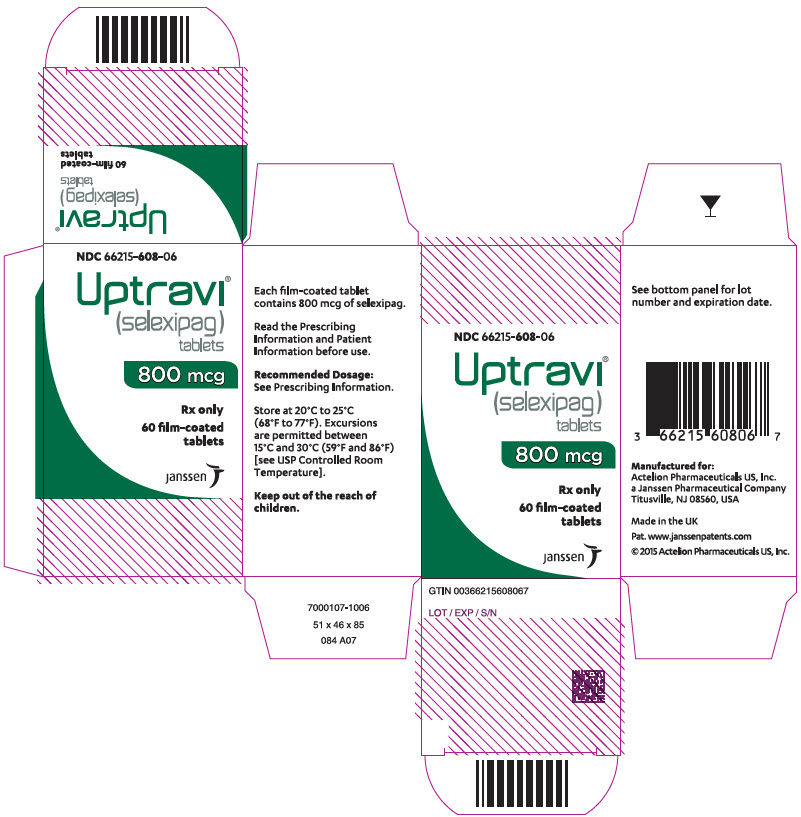
Principal Display Panel - 1000 Mcg Tablet Bottle Carton
NDC 66215-610-06
Uptravi ¬ģ (selexipag) tablets
1000 mcg
Rx only
60 film-coated tablets
janssen
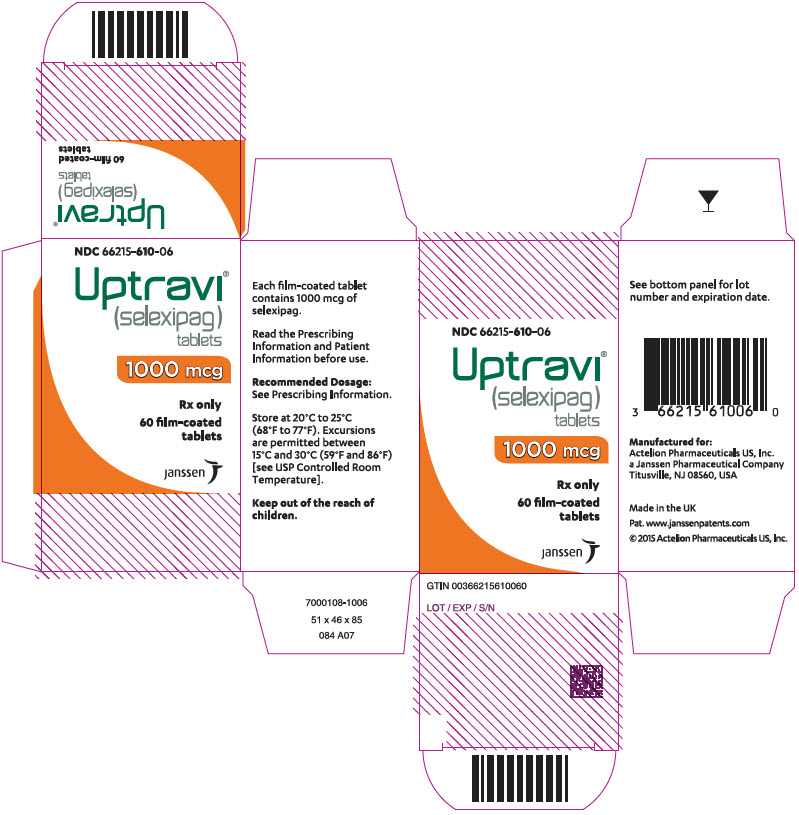
Principal Display Panel - 1200 Mcg Tablet Bottle Carton
NDC 66215-612-06
Uptravi ¬ģ (selexipag) tablets
1200 mcg
Rx only
60 film-coated tablets
janssen
Principal Display Panel - 1400 Mcg Tablet Bottle Carton
NDC 66215-614-06
Uptravi ¬ģ (selexipag) tablets
1400 mcg
Rx only
60 film-coated tablets
janssen
Principal Display Panel - 1600 Mcg Tablet Bottle Carton
NDC 66215-616-06
Uptravi ¬ģ (selexipag) tablets
1600 mcg
Rx only
60 film-coated tablets
janssen
Principal Display Panel - Kit Carton
NDC 66215-628-20
TITRATION PACK
Uptravi ¬ģ (selexipag) tablets
200 mcg
Rx only
140 film-coated tablets
Uptravi ¬ģ (selexipag) tablets
800 mcg
Rx only
60 film-coated tablets
janssen

Principal Display Panel - 1,800 Mcg Vial Carton
NDC 66215-718-01
Uptravi ¬ģ (selexipag) for injection
1,800 mcg/vial
FOR INTRAVENOUS INFUSION ONLY Reconstitute and Dilute Prior to Use.
Single-dose vial. Discard unused portion.
Rx only Sterile
One Vial
OPEN HERE

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
